第一性原理计算建模教程(基于QE)
1. 晶体结构要点
首先,简要地说明以下几个概念,以统一本文中的术语:单元,原胞,晶胞,超胞,布拉伐格子,晶面,布里渊区。
晶体(crystal),是原子、离子或分子周期性排列的结构。
格子(lattice),是数学上的点构成的周期性结构。
格子中的点,连接成平行六面体,称为单元(cell),单元具有三维周期性,是平面波程序计算的对象。
群,在集合上封闭的运算,运算满足结合律,存在单位元,存在逆元,定义为群。
可以将空间中的格子坐标看成集合,格子在空间无限延伸,如果对其进行特定的转动、平移等操作,操作前后格子坐标的集合不变,符合群的公理,则这些操作可以作为群的运算。
如果只允许平移,称为平移群。
如果只允许转动(含空间反演、镜像),称为点群。3维空间中的晶体点群有32种。
如果允许转动和平移的复合操作,称为空间群。3维空间中的晶体学空间群有230种。指定了空间群的类型,我们只需要知道在空间群操作下不重复的原子位置,就可以确定晶体结构,这些不重复的位置称为Wyckoff位置,QE输入有space_group和ATOMIC_POSITIONS { crystal_sg }来专门设置。
保持平移对称性的最小单元是原胞(primitive cell)。
保持平移对称性和点群对称性的最小单元是晶胞(unit cell)。
有时计算需要选取一个比最小单元更大的单元,习惯上称为超胞(supercell)。
在特定的平移、旋转操作下,晶体保持不变,这种在某种操作下不变的性质称之为体系的对称性,不同的操作定义了不同的对称性,例如,沿$\vec a$方向平移1个、2个、3个……平移基矢等,这些不同的对称性,一般说来是独立的,不能互相推导。
体系的薛定谔方程,由于体系的对称性,也具有变换下不变的性质,于是有特定的量子数来标记这些本征态,晶体平移对称性是一系列准连续的k值所标记的,k点所在空间称为k空间,k空间是基于特定的单元定义的,晶体的能带、声子色散等,是基于特定的k空间定义的,能带、声子色散的计算一般是基于原胞的k空间。
k空间也具有周期性,取原点周围的魏格纳-塞茨原胞,称为第一布里渊区。
布拉伐格子是按照基元+格子的概念定义的,确定布拉伐格子应满足:(1)所选平行六面体必须充分反映出格子的点群与平移群,即平行六面体必须与整个格子的晶系特征一致。(2)所选择平行六面体各个棱之间夹角为直角的数目最多,不为直角者尽可能地接近直角。(3)在满足上述(1)(2)条件后,所选择的平行六面体的体积应为最小。布拉伐格子即为晶胞。3维空间的布拉伐格子有14种。图1是14种布拉伐格子的基矢a,b,c及夹角$\alpha, \beta, \gamma$所具有的特定关系。

晶面是由至少三个不共线的格点确定的平面。
晶面一般用密勒指数标记,密勒指数是不过原点的平面在某一组基矢量方向的截距倒数之比,并约化成一组互质的整数比(详见5.1节)。这里的基矢量一般选为晶胞的基矢量,即晶面是基于晶胞定义的。
按照晶体具有的点群分类,分为7种晶系(crystal system),即:triclinic, monoclinic, orthorhombic, tetragonal, trigonal, hexagonal和cubic。
14种布拉伐格子,分为7种格点系(lattice systems),即:triclinic, monoclinic, orthorhombic, tetragonal, rhombohedral, hexagonal和cubic 注4。
2. QE中的结构定义
QE输入文件的总体结构如下图,输入文件的前半部分满足Fortran语言的语法,变量名不区分大小写,但要注意拼写是否正确。QE输入文件有CONTROL,SYSTEM,ELECTRONS等3个必需的名称列表(namelist,由Fortran语言定义,在’&’和’/’之间定义变量),以及IONS和CELL等可选的名称列表,各个名称列表的顺序是固定的,不能修改顺序;输入文件的后半部分是固定格式的ATOMIC_SPECIES,ATOMIC_POSITIONS,K_POINTS以及可选的CELL_PARAMETERS等部分,可以修改顺序。与晶体结构定义有关的内容包括:SYSTEM名称列表的变量ibrav,celldm,A,B,C,cosAB,cosAC,cosBC,nat,ntyp以及ATOMIC_POSITIONS和CELL_PARAMETERS共三个部分。

QE结构设置的种类总结如表1,单元有6种设置方法,原子坐标有4种设置方法,一共有24种组合方式(除了通过空间群设置以外)。从任意一种可以推出其余的23种,转换工具见QEtk的pw.x输入格式互换工具。
|
单元设置
|
ibrav=0 |
CELL_PARAMETERS( alat ) |
celldm(1) |
|
A |
|||
|
CELL_PARAMETERS( bohr ) |
|||
|
CELL_PARAMETERS( angstrom ) |
|||
|
ibrav≠0 |
celldm(1:6) |
||
|
A,B,C,cosAB,cosAC,cosBC |
|||
|
原子坐标设置 |
ATOMIC_POSITIONS (alat) |
||
|
ATOMIC_POSITIONS (bohr) |
|||
|
ATOMIC_POSITIONS (angstrom) |
|||
|
ATOMIC_POSITIONS (crystal) |
|||
2.1 单元的定义
QE计算的结构总是具有三维空间周期性的,需要定义周期性的单元(cell)作为计算的对象,这里的单元可以是原胞、晶胞或超胞。在QE内部用三个矢量$\vec{v_{1}},\vec{v_{2}},\vec{v_{3}}$定义单元。
\(\vec{v_{1}}=(v_{11},v_{12},v_{13}),\vec{v_{2}}=(v_{21},v_{22},v_{23}),\vec{v_{3}}=(v_{31},v_{32},v_{33})\),
单元的定义涉及到空间直角坐标系(笛卡尔坐标系)的选取,对于ibrav$\neq$0是在QE程序内部进行定义的(参见表2),用户不需要设置,也不用写出CELL_PARAMETERS;对于ibrav=0是用户通过写出CELL_PARAMETERS而确定了空间直角坐标系的。
(方法1) 设置ibrav=0,这时需要在输入文件中写入CELL_PARAMETERS,即单元的基矢量$\vec{v_{1}},\vec{v_{2}},\vec{v_{3}}$的直角坐标,这里空间直角坐标系的选法有一定的任意性,用户可以根据习惯选择,建议是右手系(虽然qe中没有强制要求右手系)。坐标的单位有三种模式供选择:alat,bohr,angstrom,其中,alat模式比较复杂,需要写celldm(1)或A,以定义alat的值,建议设置celldm(1)或A为晶胞的第一个矢量长度,即具有晶格常数的意义注1注2;bohr和angstrom模式分别是单元基矢量坐标以玻尔和埃为单位,此时不要设置celldm(1)或A。
设置ibrav=0并写出CELL_PARAMETERS这种方法适合用来设置超胞、slab模型等,也可以用来建原胞,是一种通用性较好的方法,并且与其他结构文件(cif,VESTA,POSCAR等)格式转换较为方便,也更方便进行后续计算。
(方法2) 设置ibrav$\neq$0,这时会生成布拉伐格子相应的原胞作为计算单元。表2列出了ibrav和celldm设置以及对应的$\vec{v_{1}},\vec{v_{2}},\vec{v_{3}}$原胞基矢量(相当于内部生成的CELL_PARAMETERS)。此时,celldm(1)是晶胞的第一基矢量的长度(注意区分原胞和晶胞),即alat注2,celldm(1)的单位是bohr(1 bohr = 0.52917720859 Angstrom);celldm(2)和celldm(3)定义的是比例b/a和c/a,是晶胞的基矢量的长度比(注意区分原胞和晶胞);celldm(4:6)是角度的余弦值,角度通常是相应晶胞基矢量夹角,但ibrav=5是例外;对于ibrav=-3,-5,-9,91,-12,-13,与相应的1~14设置相比,分别代表不同的空间直角坐标系的取法,或不同的单元基矢量取法;ibrav$\neq$0中的简单格子(ibrav=1,4,6,8,12,14)也可以用来设置超胞等结构(对于超胞不建议再使用ibrav=2,3,7,9,10,11,13,即面心、体心、底心等非简单格子以及ibrav=5菱方格子)。
需要说明的是,对于任意的单元,总可以建立直角坐标系,写出这个单元的CELL_PARAMETERS,即ibrav=0格式,但是,对于布拉伐格子是1~13,还存在某些单元无法写成ibrav=1~13的形式,例如将fcc的原胞基矢量中的一个反向,得到的基矢量夹角与ibrav=2的基矢量间夹角不同,不能通过坐标系变换变成ibrav=2的单元。
换句话说,ibrav=1~13模式对于除三斜外的布拉伐格子的单元定义是不完备的。好在,对于布拉伐格子是1~13,但是不能写成ibrav=1~13的情况,在周期性边界条件下总是可以避免的,根据单元的平移对称性,可以将这个单元转换成符合ibrav=1~13的单元形式,单元的基矢量坐标可以通过坐标系的旋转、坐标轴的部分反演或置换满足ibrav=1~13相应的程序内部的CELL_PARAMETERS形式,见3.1节所述的单元之间的变换。
(方法3) 设置ibrav$\neq$0,根据表2给出晶格的基矢长度和夹角的余弦,即A, B, C, cosAB, cosAC, cosBC中的部分或全部,方法3和方法2基本一样,不同的是A,B,C单位是Angstrom,并且B,C是长度而不是比值。
当然,对于方法2和方法3,设置ibrav=14,并写出全部的六个参数也是一种通用的做法。
2.2 原子坐标的定义
在定义了单元之后,用ATOMIC_POSITIONS定义单元中原子的坐标。ATOMIC_POSITIONS的单位有以下可供选择{ alat | bohr | angstrom | crystal | crystal_sg },其中,crystal是指以$\vec{v_{1}},\vec{v_{2}},\vec{v_{3}}$为基矢量的分数坐标,$\vec{X}=(x_{1},x_{2},x_{3})^{T}=x_{1}\vec{v_{1}}+x_{2}\vec{v_{2}}+x_{3}\vec{v_{3}}$。如果选择{ alat | bohr | angstrom},则原子坐标是空间直角坐标,由于结构的周期性,这里的空间直角坐标系的选择是任意的,同时与单元的空间直角坐标系也是独立选取的,但是习惯上还是与单元的空间直角坐标系保持一致,坐标值在CELL_PARAMTERS所定义的平行六面体内部。{crystal_sg}是在指定了空间群之后,定义对称性不等价的原子位置,与space_group, uniqueb, origin_choice, rhombohedral配套使用。
2.3 小结
QE在结构定义上采用了多种方式完成同一件任务的设计风格,为具有各种习惯的用户提供了得心应手的工具,但是对于初学者来说,难免有一种眼花缭乱的感觉,考虑到后处理、可视化等因素,这里推荐的方法:
(1)设置ibrav$\neq$0,对于原胞用相应的ibrav类型,对于超胞用相应简单格子的ibrav,写出celldm(1:6),这时不写CELL_PARAMETERS,输出会内部生成CELL_PARAMETERS以alat(celldm(1))为单位。VESTA画图时用输出里的CELL_PARAMETERS,需要转换单位。转为POSCAR格式可以用QEtk的P2P工具。
(2)设置ibrav=0,写出以Angstrom为单位的CELL_PARAMETERS (angstrom),对于原子坐标建议使用分数坐标,即写成ATOMIC_POSITIONS (crystal),不设置celldm(1),这时,alat和celldm(1)由程序内部设置成v1的长度,以bohr为单位。用VESTA画图时,此时CELL_PARAMETERS已经是Å为单位,转格式也比较方便。
第二种设置ibrav=0后续处理时要注意pp.x输出电荷等文件是以alat为单位输出CELL_PARAMETERS的,而与输入文件的单位不一样。vc-relax计算的最终结构是以ibrav=0搭配CELL_PARAMETERS (angstrom)的格式输出的。要注意基矢和原子坐标的有效数字位数要写得多一些,以找到正确的对称性。ibrav=0一个不足之处是输出了点群操作但是没有输出点群名称(需设置verbosity='high'),可以将qe_release_6.4/PW/src/summary.f90第608行IF ( ibrav == 0 ) RETURN加注释,重新编译。
最后,强烈建议做好结构之后,用可视化的软件如QEtk或者VESTA、Xcrysden、MS等画出晶体结构,检查一下原子间距、键角等是否正确,这些软件并不都支持QE的输入格式,需要转换格式,这时用ibrav=0也比较有利手动转格式(软件支持自动转格式当然更方便)。手动转格式的方法:例如用VESTA画图,转为POSCAR格式,输入文件拷贝CELL_PARAMETERS后面的三行作为POSCAR的第3-5行(POSCAR第二行设置为1.0),拷贝ATOMIC_POSITIONS (crystal)后面的坐标后三列,作为POSCAR里的Direct坐标,QE输出转POSCAR同上。
3. 单元变换
3.1 一般的单元变换
首先,定义一般的周期性单元的变换。按照文献[1]的约定,将(分数)坐标写为列矢量,基矢量$\vec{a},\vec{b},\vec{c}$也各为列矢量。点X在基矢$O,\vec{a},\vec{b},\vec{c}$($O$为原点)下的坐标$(x_{1},x_{2},x_{3})^{T}$定义为 \(\vec{X}=x_{1}\vec{a}+x_{2}\vec{b}+x_{3}\vec{c} =(\vec{a},\vec{b},\vec{c})\quad \begin{pmatrix} x_{1} \\x_{2} \\x_{3} \end{pmatrix} \quad\)。
考虑晶格静止不动,选择不同的基矢,即选取不同的单元,同一个点X对新的基矢$O’,\vec{a’},\vec{b’},\vec{c’}$有坐标 \(\vec{X'}=(x'_{1},x'_{2},x'_{3})^{T}=x'_{1}\vec{a'}+x'_{2}\vec{b'}+x'_{3}\vec{c'}\)。下面给出有撇号和无撇号的基矢选择下基矢和坐标的变换关系。
周期性单元的变换是保持晶格周期性的仿射变换(一般的仿射变换不保证具有晶格周期性),可以分解为线性部分和平移两个部分。
线性部分包括基矢方向和长度的改变,由一个矩阵$\mathbf{P}$表示。\((\vec{a'},\vec{b'},\vec{c'})=(\vec{a},\vec{b},\vec{c})\mathbf{P}=(\vec{a},\vec{b},\vec{c})\quad \begin{pmatrix} P_{11}& P_{12} &P_{13} \\ P_{21}& P_{22} &P_{23} \\ P_{31}& P_{32} &P_{33} \\ \end{pmatrix} \quad\\ =(P_{11}\vec{a}+P_{21}\vec{b}+P_{31}\vec{c}, P_{12}\vec{a}+P_{22}\vec{b}+P_{32}\vec{c}, P_{13}\vec{a}+P_{23}\vec{b}+P_{33}\vec{c} )\)。
知道了矩阵$\mathbf{P}$和逆矩阵$\mathbf{Q}=\mathbf{P}^{-1}$,就可以进行单元之间的变换。
3.2 晶胞和原胞的相互转换
原胞是保持平移对称性的最小单元,所以,在计算能带、声子色散时,研究对象是原胞(声子的有限位移方法需要超胞,但是,这时的超胞是一个辅助系统,声子色散仍然是基于原胞定义的)。用比原胞更大的单元计算能带会造成能带折叠(band folding),即布里渊区变小或形状发生变化,从超胞到原胞的能带可以通过能带反折叠(band unfolding)还原到原胞的能带。
文献通常按照以下约定:晶胞是定义晶面、晶向、超胞的参照,而不是原胞或其他单元。
常见的晶胞转换为原胞的变换矩阵由表3给出,反之交换$\mathbf{P}$和$\mathbf{Q}$得到。注意这里的矩阵选取并不是唯一的,这里选择的原胞基矢的原点和晶胞基矢的原点是重合的,即没有平移。
![]()
对于菱方布拉伐格子,见注4的7种空间群,cif生成的是六方的晶胞,体积是菱方的3倍,原胞计算用(ibrav=5,同时定义celldm(1)和celldm(4)),用ibrav=0时也存着晶胞转原胞的问题,转换矩阵如下,参见注3 : \(H \rightarrow R \\ P=\quad \begin{pmatrix} 2\over 3 & -{1 \over 3} & -{1 \over 3} \\ 1 \over 3 & 1 \over 3 & -{2 \over 3} \\ 1 \over 3 & 1 \over 3 & 1 \over 3 \\ \end{pmatrix} \quad Q=\quad \begin{pmatrix} 1 & 0 & 1 \\ -1 & 1 & 1 \\ 0 &-1 & 1 \\ \end{pmatrix}\)
周期性单元的变换,还可以包含平移,平移$\vec{p}$用变换前的基矢定义为:$\vec{p}=p_{1}\vec{a}+p_{2}\vec{b}+p_{3}\vec{c}$。平移的逆变换$\vec{q}=q_{1}\vec{a’}+q_{2}\vec{b’}+q_{3}\vec{c’}$,有$\vec{q}=-\mathbf{P}^{-1}\vec{p}$。
分数坐标的变换公式为: \(\quad \begin{pmatrix} x^\prime_{1} \\ x^\prime_{2} \\ x^\prime_{3} \end{pmatrix} \quad=\mathbf{Q} \quad \begin{pmatrix} x_{1} \\ x_{2} \\ x_{3} \end{pmatrix} \quad+\vec{q}\),
将晶胞转换为原胞,因为晶胞和原胞中的原子个数不同,坐标变换后会有重复或相差一个原胞格子,需要将重复的删去。反之,将原胞转换为晶胞,需要将原胞格子重复足够大以覆盖晶胞,重复后的原子分别变换到新的分数坐标,最后保留晶胞内部的原子(分数坐标在0到1之间)。
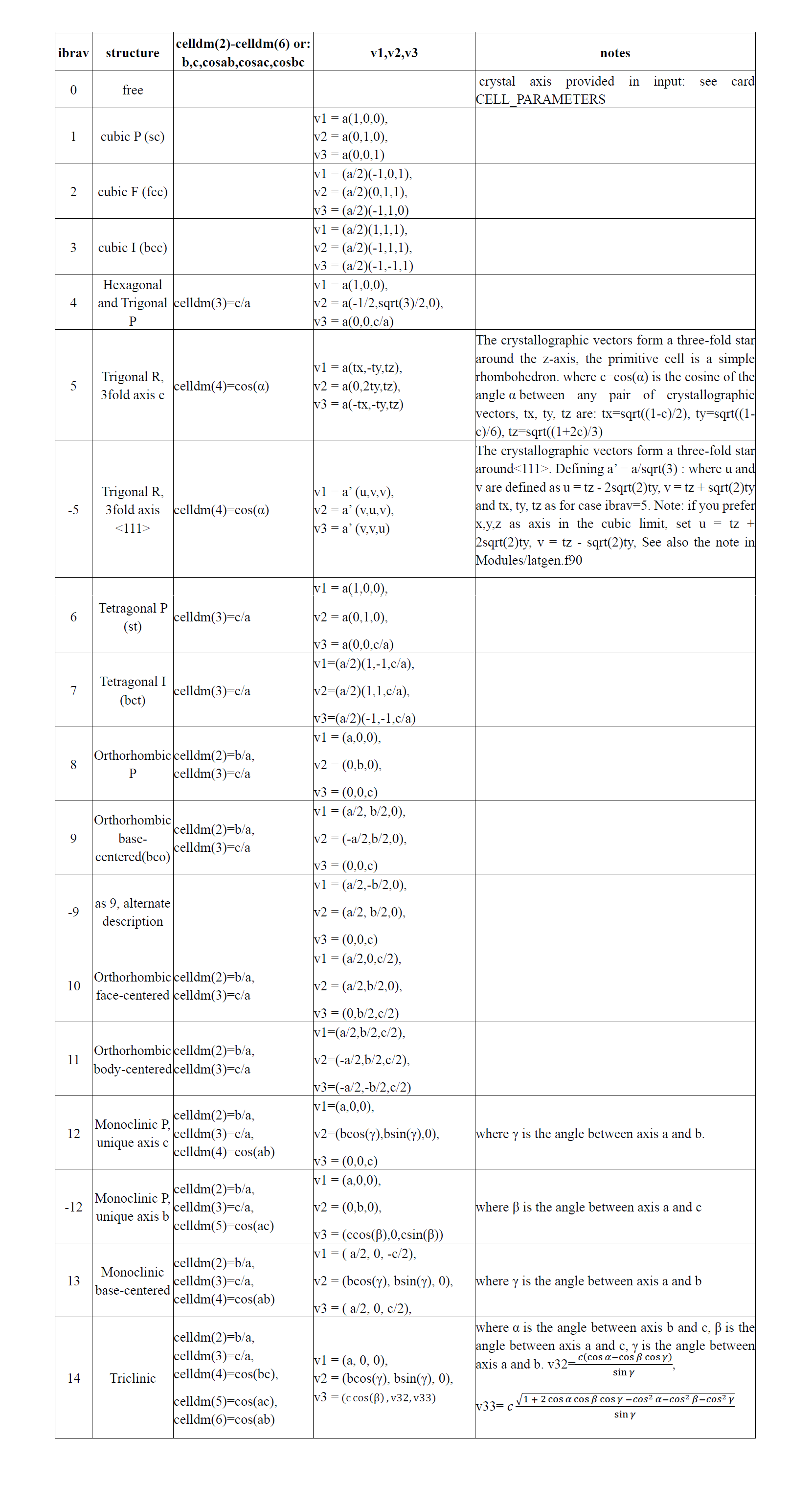
以底心单斜碳的同素异形体为例[4],用SlabMaker变换单元和原子坐标,并用VESTA验证。
先下载cif文件,用VESTA打开,画出晶胞如下:
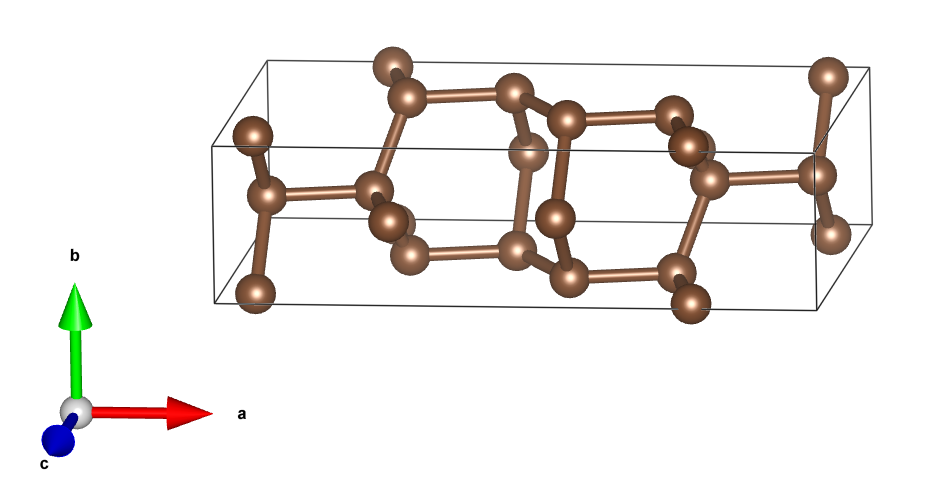
输入转换矩阵:
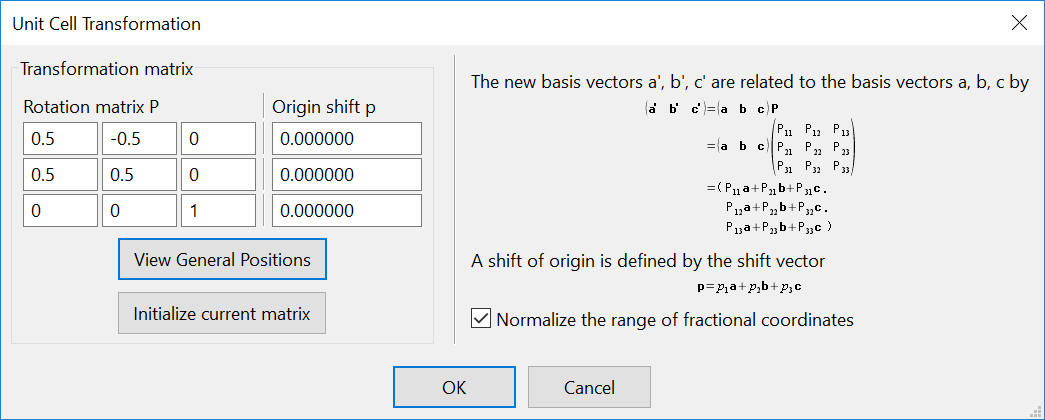
得到原胞:
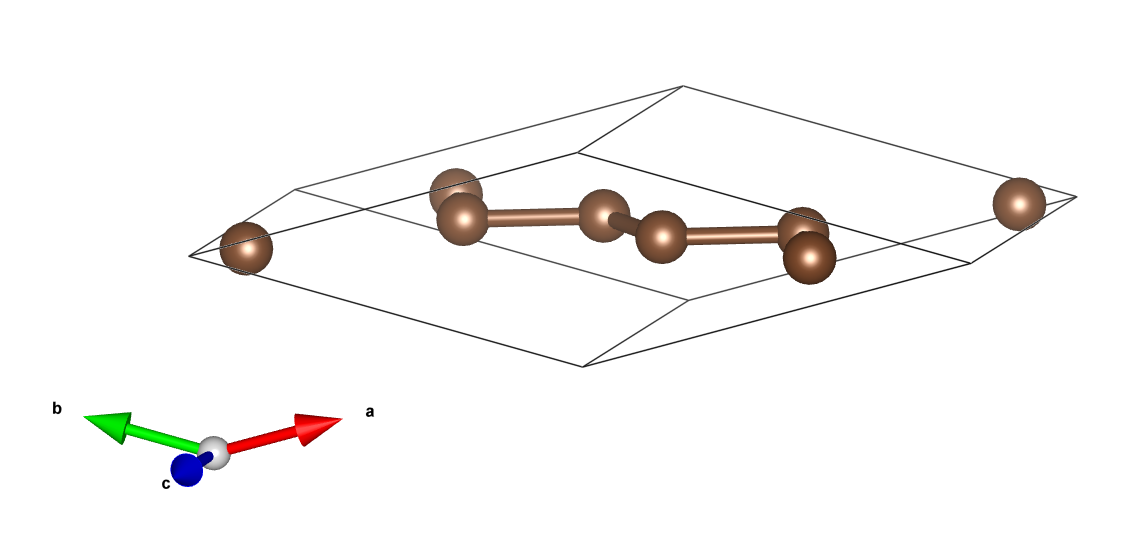
原胞和晶胞的比较(见aflow):
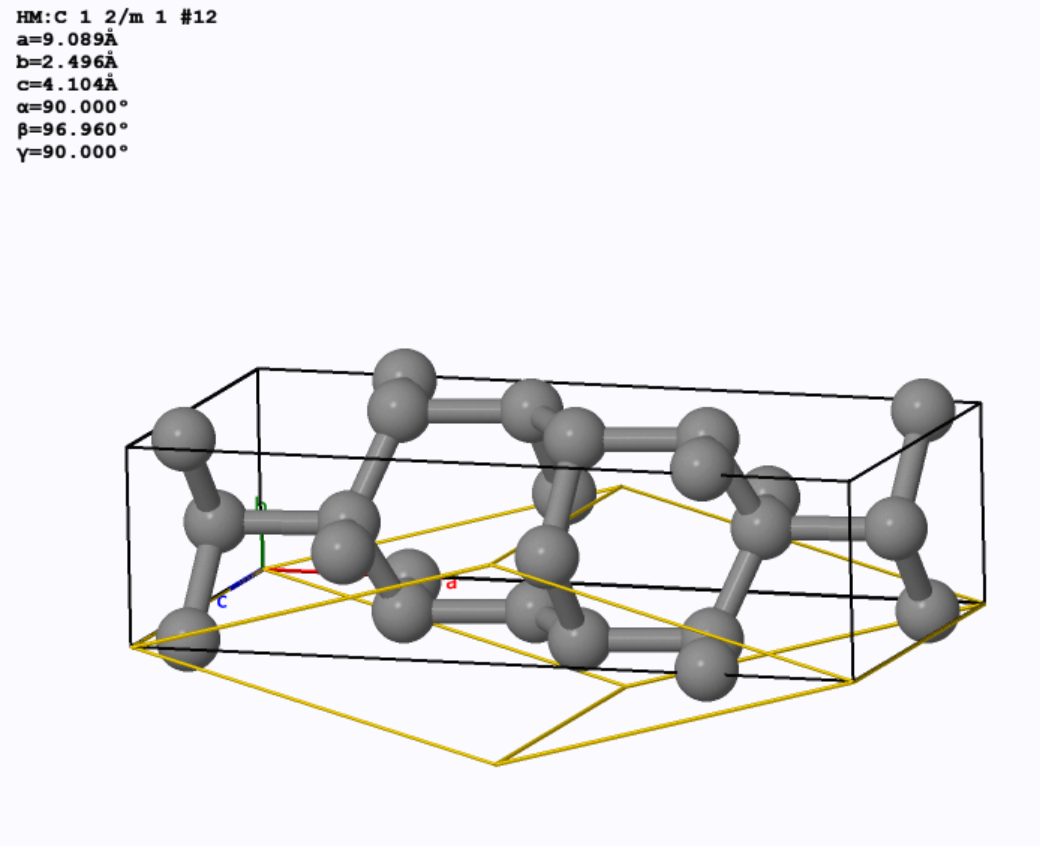
4. 分数坐标和直角坐标的相互转换
记单元基矢量为 \(\quad \begin{pmatrix} a_{1} \\a_{2} \\a_{3} \end{pmatrix} \quad\) , \(\quad \begin{pmatrix} b_{1} \\b_{2} \\b_{3} \end{pmatrix} \quad\) , \(\quad \begin{pmatrix} c_{1} \\c_{2} \\c_{3} \end{pmatrix} \quad\) .
倒格子矢量为 \(\quad \begin{pmatrix} a^{*}_{1} &a^{*}_{2} & a^{*}_{3} \end{pmatrix} \quad\) , \(\quad \begin{pmatrix} b^{*}_{1} & b^{*}_{2} & b^{*}_{3} \end{pmatrix} \quad\) , \(\quad \begin{pmatrix} c^{*}_{1} & c^{*}_{2} & c^{*}_{3} \end{pmatrix} \quad\) .
倒格子的“晶体学定义”为: $\vec{a^{*}} = { {\vec{b} \times \vec{c}} \over {V} }, $ $\vec{b^{*}} = { {\vec{c} \times \vec{a}} \over {V} }, $ $\vec{c^{*}} = { {\vec{a} \times \vec{b}} \over {V} }, $
其中单元体积$V= \vec{a} \cdot (\vec{b} \times \vec{c})$,这里的体积V当$\vec{a}, \vec{b}, \vec{c}$构成右手系时为正,否则为负。
可以证明,倒格子矩阵是单元基矢量(写成列矢量)矩阵的逆矩阵。
\(\quad \begin{pmatrix} a^{*}_{1} & a^{*}_{2} & a^{*}_{3} \\ b^{*}_{1} & b^{*}_{2} & b^{*}_{3} \\ c^{*}_{1} & c^{*}_{2} & c^{*}_{3} \end{pmatrix} \quad \quad \begin{pmatrix} a_{1} & b_{1} & c_{1} \\ a_{2} & b_{2} & c_{2} \\ a_{3} & b_{3} & c_{3} \end{pmatrix}\quad =I\) .
[还有一种倒格子的“物理学定义”,也是QE中使用的定义,等号右边多一个2$\pi$,为了后面的叙述方便,这里不采用这种定义, $\vec{a^{*}} = 2 \pi { {\vec{b} \times \vec{c}} \over {V} }, $ $\vec{b^{*}} = 2 \pi { {\vec{c} \times \vec{a}} \over {V} }, $ $\vec{c^{*}} = 2 \pi { {\vec{a} \times \vec{b}} \over {V} }$]。
可以将3.1节的单元变换矩阵用在直角坐标和分数坐标的转换,将直角坐标看成单位正交基矢对应的分数坐标。这时,单元基矢(列矢量)组成的矩阵即为单位正交基矢到单元的变换矩阵,而逆矩阵即为倒格子基矢量组成的矩阵(行矢量)。
对于单元中的原子,记原子的分数坐标为$(x_{1},x_{2},x_{3})^{T}$,直角坐标为$(z_{1},z_{2},z_{3})^{T}$,采用倒格子的晶体学定义。分数坐标转为直角坐标有:
\(\quad \begin{pmatrix} z_{1} \\ z_{2} \\ z_{3} \end{pmatrix} \quad = \quad \begin{pmatrix} a_{1} & b_{1} & c_{1} \\ a_{2} & b_{2} & c_{2} \\ a_{3} & b_{3} & c_{3} \end{pmatrix}\quad \begin{pmatrix} x_{1} \\ x_{2} \\ x_{3} \end{pmatrix}\) .
直角坐标转为分数坐标有:
\(\quad \begin{pmatrix} x_{1} \\ x_{2} \\ x_{3} \end{pmatrix} \quad = \quad \begin{pmatrix} a^{*}_{1} & a^{*}_{2} & a^{*}_{3} \\ b^{*}_{1} & b^{*}_{2} & b^{*}_{3} \\ c^{*}_{1} & c^{*}_{2} & c^{*}_{3} \end{pmatrix}\quad \begin{pmatrix} z_{1} \\ z_{2} \\ z_{3} \end{pmatrix}\) .
5. 非周期性系统
分子、团簇、纳米晶体、具有点缺陷的固体等不具有周期性,纳米线是1维周期性的,固体表面、量子阱、二维材料等是2维周期性的,QE这样的基于平面波基函数的程序,对于这些非三维周期性的材料需要采取超胞近似,即选取足够大的周期单元,在有必要时,还需要在单元中的一部分空间不加入任何原子,也就是引入真空,以隔离非周期性的维度。真空的厚度,要让两个表面的作用可以忽略,同时,考虑可接受的计算量。QEtk中的SlabMaker工具,对于平板模型,真空层推荐值是15Å。
5.1 平板(slab)模型的建立
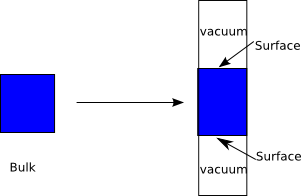
对于固体表面,平面波计算要首先建立平板模型,选取垂直晶面方向足够厚的平板,并且加入足够厚的真空,以消除表面之间的作用,实现表面性质的计算。对于异质结构,如超晶格,需要建立repeated-slab模型,二维材料异质结,如双层石墨烯“魔角”,模型建立也会遇到有共性的问题。
对于有重构(也叫再构,reconstruction,是指表面原子发生面内平移对称性的变化)的晶体表面,要按照重构截取面内的单元,重构原子的坐标要按照实验结构或经验手动设置,这是因为如果初始位置与要研究的重构表面如果相差较大,即使原子个数相同,弛豫的结果也可能是某个亚稳态结构,所以通过relax计算不能保证弛豫到要研究的特定重构表面结构。
对于没有重构的晶体表面,需要考虑如下:
首先,确定要计算的晶面。晶面用密勒指数标记,密勒指数是不过原点的平面在基矢方向的截距倒数约化成的整数比,注意密勒指数是相对晶胞定义的,通常是三个数,如(001),(110), (531)等等,一般低密勒指数的面较常见,但实验上也会出现高密勒指数的面。对于六方和三方(菱方)晶体,习惯上用四个数的密勒指数,如(0001),(1$\overline{1}$01)等,这里是选了垂直三重旋转轴的面内3个基矢以及沿着三重旋转轴的1个基矢,一共4个基矢而定义的密勒指数,其中,前三个指数的和一定是零,所以只有3个独立的指数,有的文献对于六方或三方结构的晶面也用三指数的表示。晶面确定后,还要根据实验确定表面原子的种类,实验上由于生长条件不同可能有多种表面原子的情况,例如GaN(0001)的表面有Ga和N原子截止的不同情况。
第二,要找到面内的最小周期性单元。先通过密勒指数的定义找到一个面内周期单元(可能非最小),以这个面内周期单位为基础,QEtk采用了了一种基于蛮力法(brute-force)的找面内最小周期单元的算法,从小到大寻找若干基矢对,对于超胞中每一个原子,分别找到面内的最小的几个单元,在不同原子取法中,找到共同面内周期单元中最小的一个,即是要找的表面结构二维最小单元,这时找到的单元并不唯一,为了减小结果的随机性,选取满足以下条件的单元:$a \le b$,夹角有90度选为90度,六方按照文献常见的取为120度,其余选为最接近90度的锐角。QEtk中的开源项目SlabMaker实现了建slab的功能,并且提供了在线工具。其他的实现,包括用MS的建模模块进行;公开的其他来源的讨论包括文献[5]。
第三,确定平板和真空的厚度。无论在平板内两个表面的距离,还是真空两边表面的距离都要足够大,以隔离两个表面的作用,模拟固体表面的性质,真空至少需要10Å到20Å。建议真空放在单元的z方向的两端(如上图,垂直表面方向记为z)。有时,为了方便,Slab模型的单元的基矢并不是正交的,但是考虑到周期性这种单元与正交单元是等价的。有的文献描述平板厚度时,提到了层(layer)的概念,层并没有无争议的定义,需要依情况而定。有时,材料在垂直晶面方向有周期性,那么层可能是周期的个数;而另一些材料有若干层原子为一组,组与组之间距离较大可以明显划分开,这里的组就是层;还有的材料,在垂直晶面方向杂乱无章,一个原子或几个具有相同z坐标的原子就是一层。
建好超胞之后,变换单元原子和分数坐标的方法为:将空间直角坐标系做旋转,总可以实现x轴沿第一个基矢(记为$\vec{a}$)方向,z轴与x轴垂直且沿第三个基矢(记为$\vec{c}$)方向(原第一和第三基矢不垂直的,由于三维周期性,也可以将第三基矢投影到垂直表面方向,从而与第一基矢垂直),首先,将第三基矢投影到垂直表面方向:
$\vec{\tilde{c}}
= (\vec a \times \vec b)\frac {\vec{c} \cdot (\vec a \times \vec b)} {\lvert\vec a \times \vec b\rvert^2}
$,
再将单元变换为:
\(\quad \begin{pmatrix} \lvert \vec a \rvert & 0 & 0 \\ {\vec a \cdot \vec b } \over {\lvert \vec a \rvert} & \sqrt {\vert\vec b\vert^{2}-({\vec{a} \cdot \vec{b}}/ {\vert\vec a\vert})^{2} } & 0 \\ 0 & 0 & \vert \vec{\tilde{c}}\vert \\ \end{pmatrix} \quad\),
真空厚度记为$d_{vacuum}$,找到原子分数坐标最大和最小的两个原子,新的z方向长度为\(\vert\vec c^{\prime}\vert=(x_{max,3}-x_{min,3})\vert\vec c\vert+d_{vacuum}\)。加入真空后,分数坐标如下变换,可以将真空置于单元的两端,
\(\vec{X^{\prime}}=(x^{\prime}_{i1},x^{\prime}_{i2},x^{\prime}_{i3})^T=(x_{i1},x_{i2},[d_{vacuum}/2+(x_{i3}-x_{min,3})\vert\vec c\vert]/{\vert\vec c^{\prime}\vert})^T\)。
下面以$\alpha-Al_{2}O_{3}$的(110)面为例,用SlabMaker建slab模型,并用VESTA画图。
从COD下载$\alpha-Al_{2}O_{3}$的cif文件,用VESTA打开,材料具有菱方的原胞,密勒指数是相对晶胞定义的,画出六方的晶胞如下:
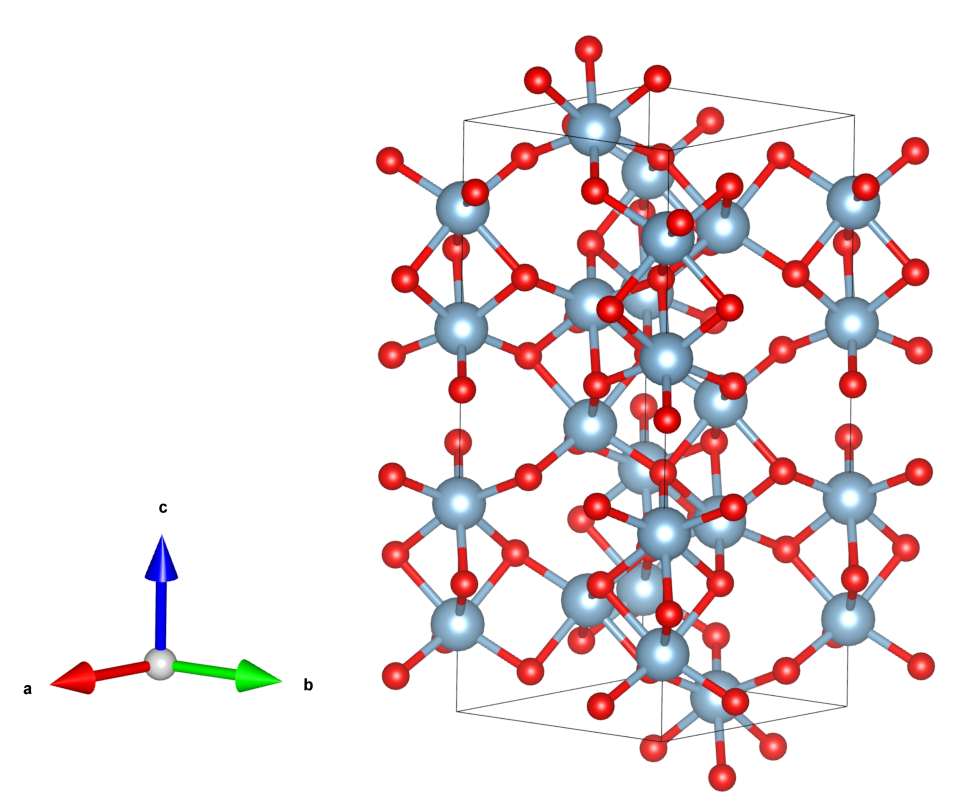
用VESTA导出POSCAR格式文件,命名为Al2O3.vasp。
从这里下载build.py文件,运行python
from build import CELL
unit=CELL("Al2O3.vasp")
slab=unit.makeslab([1,1,0], layer=2)
slab.print_poscar("./tmp/slab.vasp")
得到变换矩阵
P1 = [[ 1. 0. 2.]
[-1. 0. 2.]
[ 0. -1. 0.]]
P2 = [[-3.33333328e-01 6.66666672e-01 0.00000000e+00]
[-3.33333313e-01 -3.33333313e-01 0.00000000e+00]
[ 3.00064645e-09 3.00064645e-09 1.00000000e+00]]
reduced slab cell
[[-2.74760979e+00 -4.33033313e+00 7.13849973e-08]
[ 5.49521970e+00 -4.33033313e+00 7.13849973e-08]
[ 0.00000000e+00 0.00000000e+00 2.37898727e+01]]
reduced slab No. of atoms: 40
slab and vacuum length: 8.78987274001554 15.0 Ang.
inplane edge and angle: 5.128464149403621 6.99637224468677 84.15650034981714 degree.
reduced slab cell area: 35.694197603259965 Ang^2.
其中变换P1是得到一个预选的单元,对预选单元加入真空,沿着垂直表面方向转动c(变换矩阵见前文),变换P2是将单元约化到具有110面内最小二维周期单元的slab,面内基矢量的夹角是84.16°,结果参考见[6]。
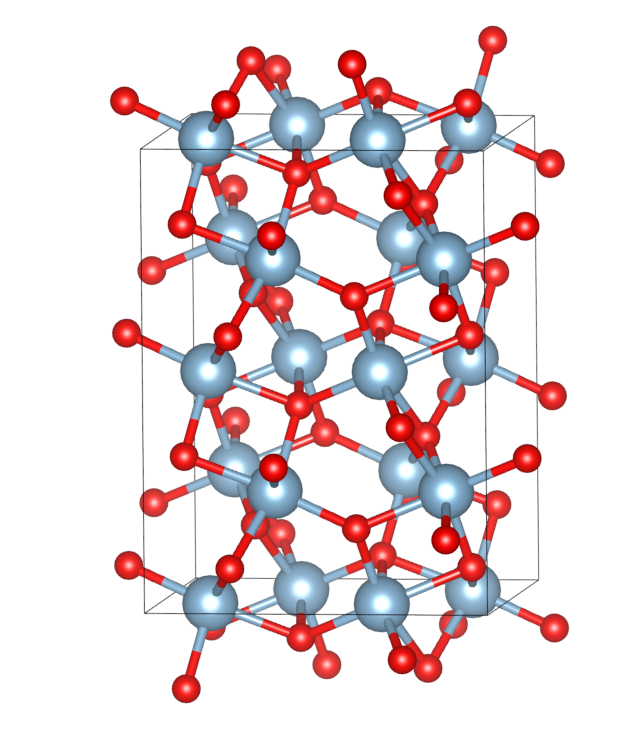
build.py输出了slab的POSCAR(真空厚度和层数在源程序中设置),见运行目录的tmp/slab.vasp。最终slab如图。
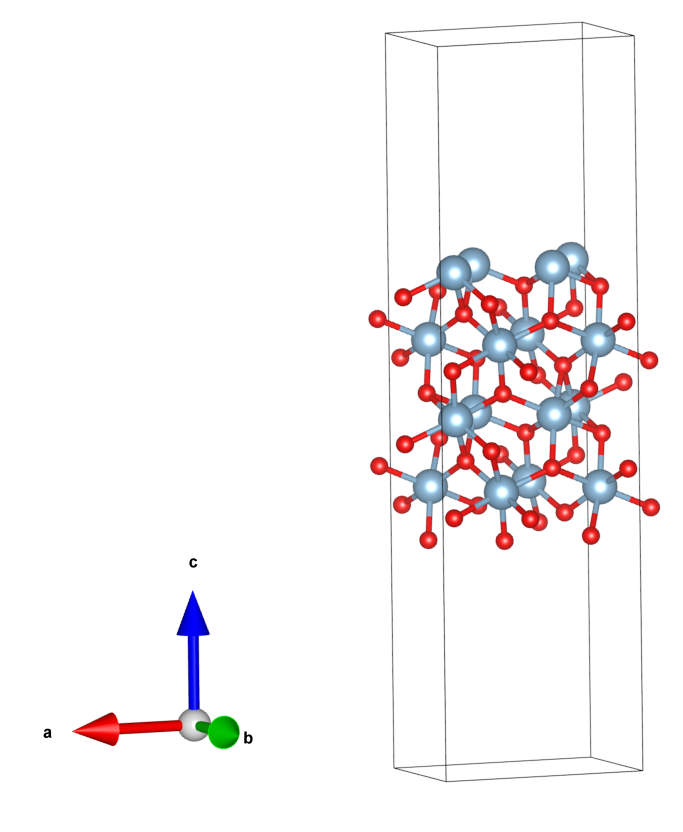
以上是slab的c方向恰好具有周期性的情况,另外一种情况则是当单元的c方向沿着表面法向时,表面法向不具有周期性(或具有极长的周期性),不同于文献[5]的做法,这里在加入真空之后,将单元的c投影到z方向,由于面内的周期性边界条件,这么做是可行的。下面以$\alpha-Al_{2}O_{3}$的(104)面为例(这与文献[5]的$\alpha-Fe_{2}O_{3}$是同一种结构)。
from build import CELL
unit=CELL("Al2O3.vasp")
slab=unit.makeslab([1,0,4], layer=1)
slab.print_poscar("./slab.vasp")
得到slab.vasp,用VESTA画图如下。
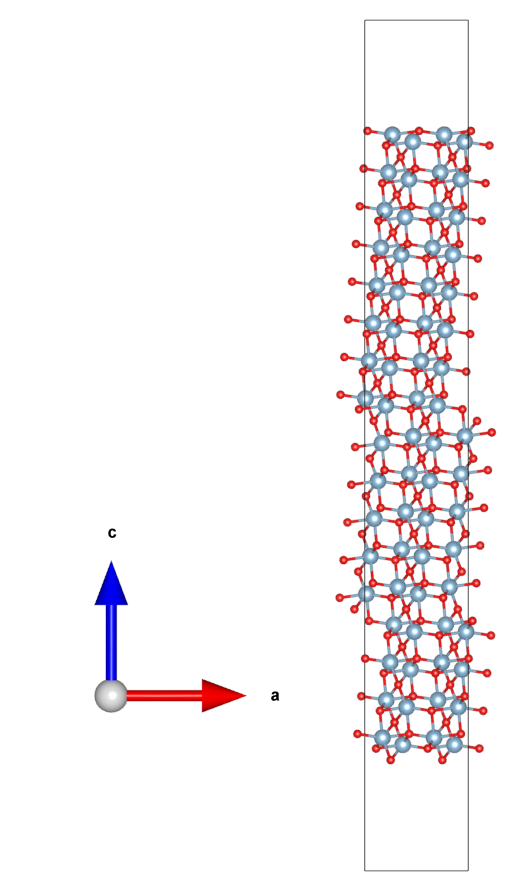
可以看到与文献[5]Fig.1(d)的面内是等价的,单元的c沿垂直表面方向有利于如功函数等的计算。建好slab之后,可以根据需要删掉部分原子以得到特定的截止表面和厚度。
注释
1.自6.4.1版本,官方不推荐celldm(1)=1.88972613(任何<2的值),即将celldm(1)设置为单位转换系数的做法,这里也修正为celldm(1)设置为晶格常数,或用ibrav$\neq$0。
2.关于alat,alat是qe内部定义的量,以bohr为单位,具有晶格常数的意义(这里晶格常数特指晶胞的第一个基矢量长度),在pw.x的输出接近开头处有 lattice parameter (alat) = x.xxxx a.u.。
(1)当ibrav=0,且设置CELL_PARAMETER{bohr或angstrom}时,alat是CELL_PARAMETER第一行矢量的长度,此时不允许写celldm,否则会和CELL_PARAMETER冲突,此时,alat对于晶胞是晶格常数,对于非简单格子的原胞则不是晶格常数;
虽然没有硬性规定,但是,建议晶胞的三个基矢量按照从小到大的顺序排列,即a≤b≤c,因此CELL_PARAMETER第一行矢量具有晶格常数的意义。
(2)当ibrav=0,且设置CELL_PARAMETER{alat}时, alat=celldm(1)或A/0.529(celldm(1)和A分别是以bohr和Angstrom为单位),这里celldm(1)或A取值有一定的任意性,但是必须要写,这里建议取为晶胞第一个基矢量的长度,即具有晶胞晶格常数的意义;
(3)对于ibrav$\neq$0,alat=celldm(1)或A/0.529,alat具有晶胞晶格常数的意义。
对于输入,使用CELL_PARAMETER {alat}、ATOMIC_POSITIONS {alat}时用到了alat,这时单元及原子坐标参数是以alat为单位的,其他情况则没有涉及到alat,可以忽略。
对于输出,pw.x有些输出量用到了alat为单位,这里就不再列举,根据情况判断。
3. 注意晶胞和原胞的区别,对于非简单格子ibrav$\neq0$适合于设置原胞(对于简单格子ibrav$\neq$0当然也是设置了原胞),布拉伐格子中的7个简单格子本身就是原胞,而且,除了菱方外的6个简单格子,不仅是原胞,同时也是晶胞,菱方的布拉伐格子是原胞但不是晶胞,菱方的晶胞是六方的简单格子,体积是原胞的3倍,而底心、面心、体心的7个布拉伐格子本身是晶胞,存在体积更小的原胞。
4. trigonal三方晶系有两种布拉伐格子,一种是ibrav=5,菱方(rhombohedral)布拉伐格子,另一种是ibrav=4,六方(hexgonal)布拉伐格子,晶体属于菱方还是六方要看具体的空间群,在hexgonal和trigonal晶系中,7个空间群($R3, R\overline{3}, R32, R3m, R3c, R\overline{3}m, R\overline{3}c$)具有菱方布拉伐格子的原胞,其余的45个空间群具有六方布拉伐格子的原胞。这里的菱方和六方是指晶体的格点系统lattice system,而非晶系,格点系统是按照布拉伐格子分类的,晶系是按照晶体点群分类的。
参考文献
-
International Tables for Crystallography (2006). Vol. A, Chapter 5.1, pp. 78–85.
-
https://en.wikipedia.org/wiki/Bravais_lattice
-
http://www.quantum-espresso.org/Doc/INPUT_PW.html
-
Q. Li et. al., Superhard Monoclinic Polymorph of Carbon, Phys. Rev. Lett. 102, 175506 (2009), doi:10.1103/PhysRevLett.102.175506.A. R. Oganov and C. W. Glass, Crystal structure prediction using em ab initio evolutionary techniques: Principles and applications, J. Chem. Phys. 124, 244704 (2006), doi:10.1063/1.2210932.
-
Wenhao Sun, Gerbrand Ceder, Efficient creation and convergence of surface slabs. Surface Science 617 (2013) 53–59.
-
Takahiro Kurita, Kazuyuki Uchida, and Atsushi Oshiyama, Atomic and electronic structures of α-Al2O3 surfaces, Phys. Rev. B 82, 155319(2010).
建模型的第一原理是符合实际。
一部大书是一项大罪。 ——卡利马科斯 (Callimachus)
Update on 2019/04/22.
Update on 2019/11/15.
Update on 2022/08/10.
本文总阅读量次