- QE实践详解
QE实践详解
1. 计算半导体SiC的能带
计算分为三个步骤:1. 优化晶格常数;2. 自洽计算;3. 能带计算。
1.1 设置结构参数
1.1.1 晶体结构信息的获取
首先介绍晶体结构信息的获取。对于材料的第一性原理计算,研究的对象是模型,对于SiC这样的晶体,模型由晶体结构信息所指定,包括原子的元素种类、原子的坐标、晶胞基矢量等信息。晶体结构信息的来源可以是cif文件,也可以是文献、手册的内容。有了这些信息,就可以写出pw.x的输入文件。关于输入文件及建模型的方法详见建模教程。
下面介绍cif文件的获取。碳化硅SiC有多种结构,这里选择了比较简单的闪锌矿(Zincblende)结构。计算需要知道SiC的原胞尺寸、原子坐标和晶格常数,这些参数一般是从实验得到的,这里推荐一个数据库Crystallography Open Database(COD,如果你手头有更好的cif数据库当然也可以)。打开COD主页,点击左侧的“search”。
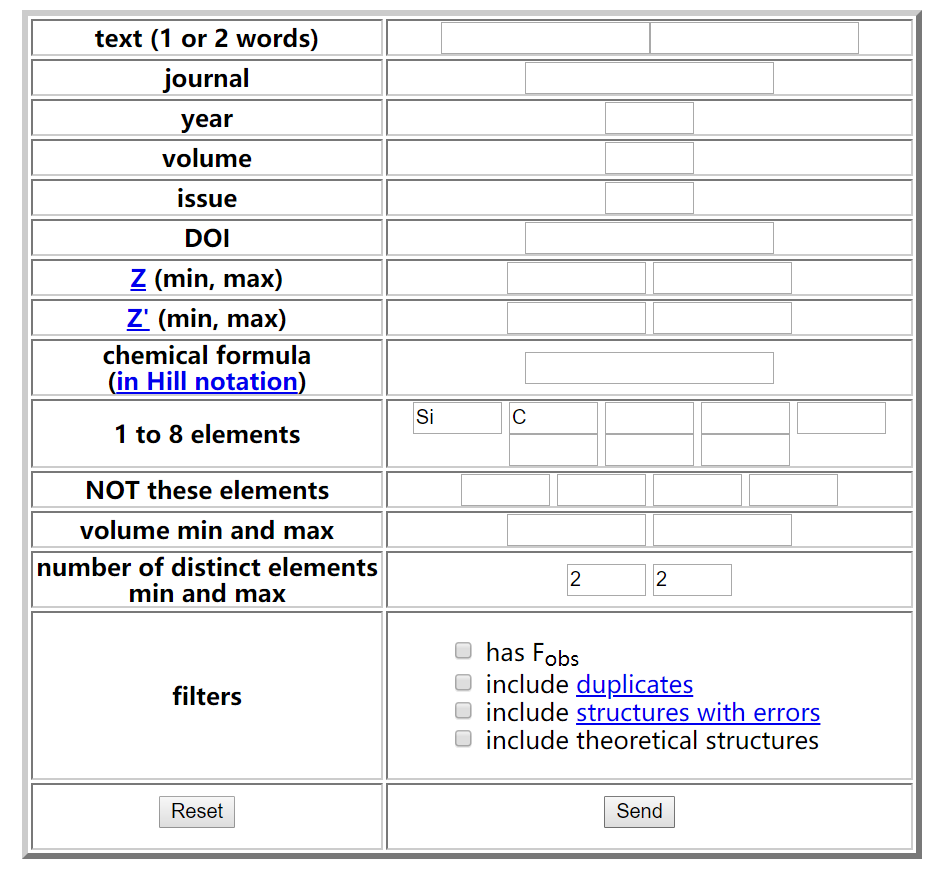
在搜索页面“1 to 8 elements”一栏输入Si和C,“number of distinct elements min and max”一栏输入2和2,表示搜索Si和C元素组成的二元化合物,点击“send”开始搜索。搜索的结果包含了数据库中的SiC二元化合物的多种组分及结构,其中有两个空间群为$F \overline{4}3M$(No. 216)是我们要找的闪锌矿结构,晶格常数略有不同,分别是4.348Å和4.358Å,选择可信度高的的一个,考虑到后面要计算晶格常数的理论值(即vc-relax计算),这里的两个选择差别不大,没有什么影响。
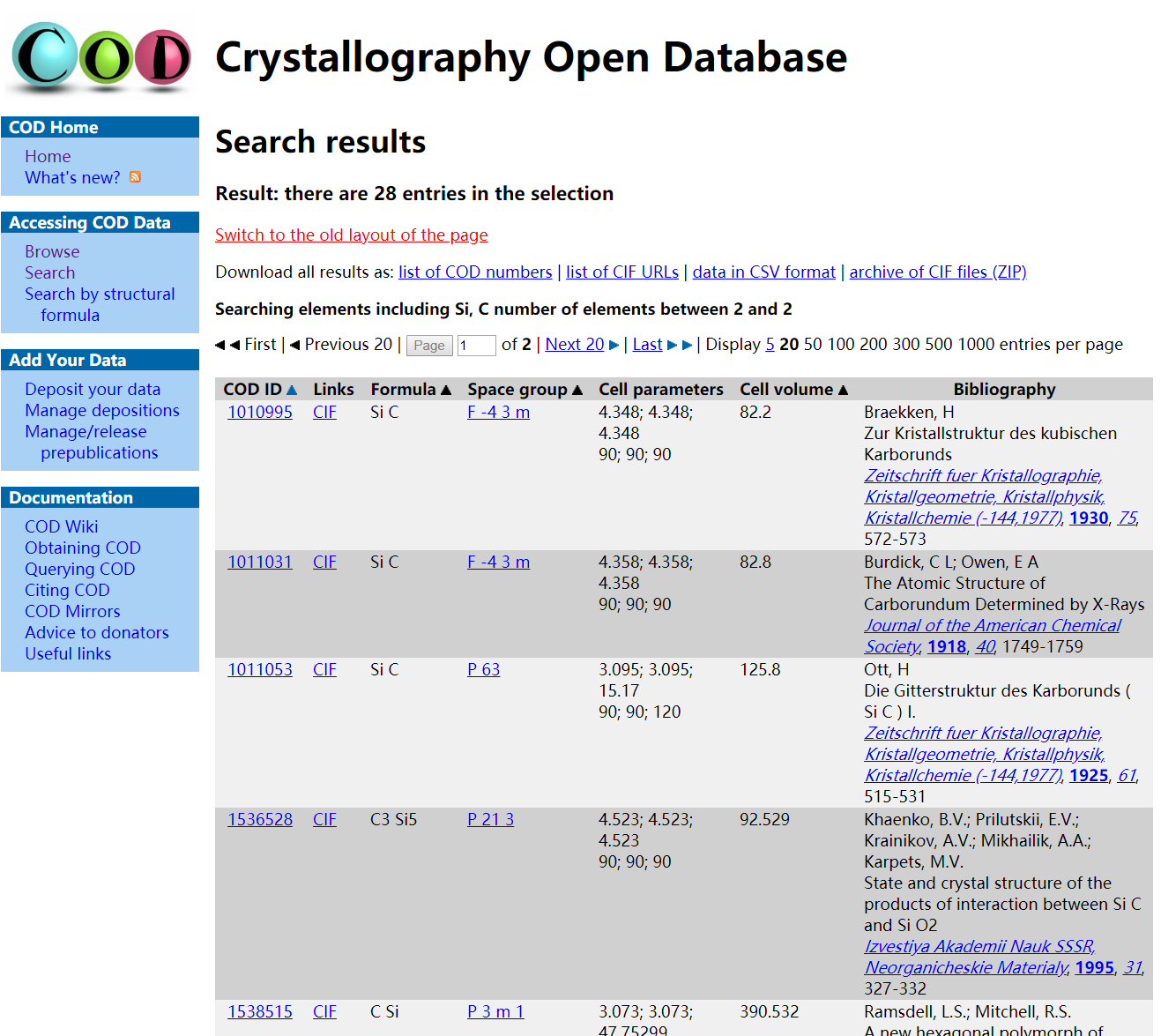
点击CIF下载后缀为cif的文件。
1.1.2 生成pw.x输入相关信息
计算能带是对材料的原胞进行计算,而cif文件一般是晶胞(少数情况下cif文件是原胞)。接下来需要从cif文件出发,得到原胞结构的pw.x输入文件,下面介绍两种流程:
(方法1) 使用VESTA或MS等工具
用VESTA打开cif文件,(或使用其他可视化程序,如Material Studio等)。在VESTA中,点击“File”——“Export Data”,选择VASP的POSCAR格式,将文件保存为“SiC.vasp”文件。
Si C
1.0
4.3480000496 0.0000000000 0.0000000000
0.0000000000 4.3480000496 0.0000000000
0.0000000000 0.0000000000 4.3480000496
Si C
4 4
Direct
0.000000000 0.000000000 0.000000000
0.000000000 0.500000000 0.500000000
0.500000000 0.000000000 0.500000000
0.500000000 0.500000000 0.000000000
0.250000000 0.250000000 0.250000000
0.750000000 0.750000000 0.250000000
0.750000000 0.250000000 0.750000000
0.250000000 0.750000000 0.750000000
上面是得到的文件(SiC.vasp),包括了cell尺寸(以Å为单位,第3-5行)和原子坐标(分数坐标,第9-16行)。需要注意的是,导入cif文件得到一般是晶胞,而晶胞对于非简单格子(面心、体心、底心等)不是原胞,为了转化为原胞,最简单的方法是借助Material Studio的建模功能,也可以参考这里表格2,得到从晶胞到原胞的转换矩阵,输入到VESTA中得到原胞,再用VESTA输出POSCAR格式。具体操作是:在VESTA中点击“Edit”——“Edit Data”——“Unit Cell”——“Transform”,输入转换矩阵如下图。
![]()
得到的结构文件如下,我们需要将其改成pw.x输入格式,见INPUT_PW。
Si C
1.0
3.0745003223 0.0000000000 0.0000000000
1.5372501612 2.6625953831 0.0000000000
1.5372501612 0.8875317944 2.5103190013
Si C
1 1
Direct
0.000000000 0.000000000 0.000000000
0.250000000 0.250000000 0.250000000
可以发现VESTA对CELL做了处理变成了一个下三角阵,但是这样不太容易找对称性(实际上pw.x确实没找全!),可以将其改写成INPUT_PW所具有的格式如下(这里是进行了直角坐标系的变化,目前只能通过观察手动进行):
system: Si C
1.0
-2.174000 0.000000 2.174000
0.000000 2.174000 2.174000
-2.174000 2.174000 0.000000
Si C
1 1
Direct
0.000000000000 0.000000000000 0.000000000000
0.250000000000 0.250000000000 0.250000000000
这样找到的对称性是正确的(24个operation)。
另外,在新版VESTA中增加了生成原胞功能。
POSCAR格式与pw.x格式转换,可以手动编写文件,当然这需要对这两种格式的规则熟悉,也可以用QEtoolkit中的P2P工具。
(方法2) 使用QEtoolkit
QEtoolkit提供了建模、格式转换、计算流程及后处理网页工具,完整支持QE格式。
使用QEtoolkit中的Cif2QE工具,将CIF文件转为pw.x输入格式,通过可视化控件查看结构。上传cif文件,点击“提交”,会生成pw.x输入文件,同样的,需要对此文件进行从晶胞到原胞的转换。
使用QEtoolkit中的Unit2Prim工具,将晶胞转换为原胞,采用了表格2所述方法,可以实现全部14种布拉伐格子的晶胞转原胞的功能,如果前面cif得到的已经是原胞或者属于简单格子,这一步当然可以省去。在Unit2Prim页面上传晶胞的pw.x输入文件,并指定Bravais格子类型,这里选择面心立方,即”2”,点击“提交”,得到原胞的输入文件。
除了Unit2Prim,也可以用QEtoolkit中的FindPrim工具,FindPrim采用了spglib提供的搜索原胞算法,对于晶胞甚至超胞都可以找到对应原胞。
作为检验和对照的方法,这里列出将晶胞转化成原胞的其他参考方法:[1]avogadro,[2]phononpy,[3]celltran,[4]Material Studio的CASTEP计算前检查原胞的功能。这里列出的方法,只做为参考,不保证它们的结果一致,也不保证它们的结果的正确性,正确性需要读者自行判断。
(方法3) 其实有经验的用户,根据cif、文献、手册的内容,直接手动写ibrav,celldm这些参数,也是完全可行的。
pw.x 输入与结构有关的包括SYSTEM部分的
ibrav,celldm,nat,ntyp
以及ATOMIC_POSITIONS和CELL_PARAMETERS两部分。
这里推荐两种写法:(1)根据布拉伐格子设置ibrav,根据晶格常数设置celldm(1-6),这时不写CELL_PARAMETERS。(2)设置ibrav=0和CELL_PARAMETERS (angstrom),这时不允许写出celldm(1),程序会自动在内部设置celldm(1) = alat = $\left | \vec v_{1} \right | = \sqrt{ \vec v_{1} \cdot \vec v_1} $ ,方便转格式用VESTA画图。
我们通过cif文件的空间群(见ITC,或通过文献),知道了SiC的布拉伐格子是面心立方,就可以设置ibrav=2,这时不需要转化为原胞,程序内部会将CELL_PARAMETERS设置为原胞的基矢量坐标;知道了晶胞的晶格常数为4.348Å,就可以设置celldm(1)=8.216529225786777,是以Bohr为单位的晶胞的基矢长度(4.348/0.52917720859);不写CELL_PARAMETERS。注意celldm(1)是晶胞第一个基矢长度而不是原胞的基矢长度(菱方除外,此时ibrav=5,celldm(1)是原胞基矢长度)。
至于原子坐标,对于熟悉的结构,比如SiC的闪锌矿结构,原胞两个原子的分数坐标分别是(0.0,0.0,0.0)和(0.25,0.25,0.25),按照格式写出ATOMIC_POSITIONS部分,坐标多写几位有效数字,有利于找到对称性。
1.1.3 小结
计算一种材料结构参数的步骤,总结起来就是:方法1,用VESTA,cif文件——晶胞POSCAR——原胞POSCAR——pw.x输入;方法2,QEtk的cif2qe工具可以将cif文件转为晶胞的pw.x输入,再用prim2unit或findprim工具转为原胞的pw.x输入;方法3,手动写。总之,过程还是有一些曲折的。
以上方法(1)(2),适用于初学者,也是自动化的通用方法,可以用在高通量计算上。
注:自6.4.1版本,官方不推荐celldm(1)=1.88972613(任何<2的值)即单位转换系数的做法,这里也修正为celldm(1)设置为晶格常数,或用ibrav$\neq$0。
1.2 结构弛豫的输入文件
pw.x处理的计算包括以下7种类型,在输入文件中用calculation设置:
‘scf’:自洽计算,self-consistent field,通过迭代的方式数值求解微分-积分方程(Kohn-Sham方程),迭代收敛以电荷的变化足够小为准,最终得到自洽电荷。
‘nscf’:非自洽计算,scf计算常在k空间的网格上进行,网格要足够密以完成k空间上的积分,在DOS等计算需要更密的$\mathbf{k}$点,这时在自洽电荷基础上,计算这些更多的$\mathbf{k}$点,nscf计算保持自洽电荷不变。
‘bands’:也是一种nscf计算,$\mathbf{k}$点按照三维k空间中的特殊路径选取。
‘relax’:一系列scf计算,通过Hellman-Feynman力计算离子坐标驰豫(通过优化算法找到受力为零的结构),relax计算时固定cell不变。
‘vc-relax’: 允许cell变化的relax,通过应力的计算改变cell。
‘md’:分子动力学,将电子对离子的作用看成离子感受到的势,根据势能和离子初始速度求解离子运动的经典力学方程。
‘vc-md’:允许cell改变的md。
pw.x的输入说明见INPUT_PW。注意默认的单位,其中原子单位制为(以下数值见源程序q-e-qe-6.3/Modules/constants.f90):
1 bohr = 1 a.u. (atomic unit) = 0.52917720859 angstroms.
1 Rydberg (Ry) = 13.60569193 eV
关于输入文件,还需要注意其中不能含有制表符等不可打印字符,这些字符可能会在excel文件复制粘贴过程中产生,并且在文本编辑器不可见,非常隐蔽。
pw.x的初始晶体结构及晶格参数通常是实验值。但是,为了后续的计算可能需要通过力和应力的弛豫(vc-relax)得到晶格参数的理论值(如果只计算体材料能带,而所关心的问题只限于电子性质而无关晶格,使用实验值也是可以的)。
在结构驰豫(calculation=’relax’)过程中,ATOMIC_POSITIONS是根据力而变化的,如果是vc-relax,原子坐标改变的同时,CELL_PARAMETERS根据应力变化(celldm在vc-relax时是不变的)。
结构弛豫的输入文件如下:
&CONTROL
calculation='vc-relax', disk_io='low', prefix='pwscf',
pseudo_dir='./', outdir='./tmp', verbosity='high'
tprnfor=.true., tstress=.true., forc_conv_thr=1.0d-5
/
&SYSTEM
ibrav= 0,
nat= 2, ntyp= 2,
occupations = 'smearing', smearing = 'gauss', degauss = 1.0d-9
ecutwfc= 50, ecutrho = 500,
/
&ELECTRONS
electron_maxstep = 100
conv_thr = 1.0d-9
mixing_mode = 'plain'
mixing_beta = 0.8d0
diagonalization = 'david'
/
&IONS
ion_dynamics='bfgs'
/
&CELL
press_conv_thr=0.1
/
ATOMIC_SPECIES
Si 28.08550 Si.UPF
C 12.01070 C.UPF
CELL_PARAMETERS (angstrom)
2.174000000 2.174000000 0.000000000
0.000000000 2.174000000 2.174000000
2.174000000 0.000000000 2.174000000
ATOMIC_POSITIONS (crystal)
Si 0.000000000 0.000000000 0.000000000
C 0.250000000 0.250000000 0.250000000
K_POINTS {automatic}
8 8 8 0 0 0
对于熟悉的闪锌矿结构,这里的CELL_PARAMETER是常见写法,与上面按照一般方法从cif获得的原胞等价。
这里的赝势文件由ld1.x生成,ld1.x的输入文件来自pslibrary1.0,推荐使用超软赝势,相比模守恒赝势有较小的截断能。赝势使用更多内容见赝势教程。程序根据赝势自动设置了交换关联泛函的类型是PBE。
pseudo_dir=’./’是赝势文件所在目录,’./’表示赝势文件在当前目录。outdir=’./tmp’是输出电荷、波函数等文件所在目录,之后的nscf等计算要读入这些文件,所以要保持一致。prefix=’pwscf’表示outdir目录下文件前缀,后续计算也要保持一致。
将输入文件保存成vc.inp,和两个赝势文件放在同一目录,运行pw.x < vc.inp > vc.out 开始计算。(并行计算,安装了openmpi则运行mpirun -np 16 pw.x < vc.inp > vc.out,其中,16是使用的核数。)
在vc-relax(relax,scf)计算时,程序会找体系的点群对称操作以减少计算量,并在弛豫时保持对称性,如果想要允许弛豫改变对称性,需设置nosym=.true.,noinv=.true.。寻找对称性时考虑了FFT格点,有可能会舍去部分晶体点群操作,有必要时需要设置nr1, nr2, nr3以允许这部分点群操作。
计算结束后,运行
awk '/Begin final coordinates/,/End final coordinates/{print $0}' vc.out
,得到以下输出(或在输出文件中可以找到)
Begin final coordinates
new unit-cell volume = 141.54631 a.u.^3 ( 20.97500 Ang^3 )
density = 3.17432 g/cm^3
CELL_PARAMETERS (angstrom)
2.188890250 2.188890250 0.000000000
0.000000000 2.188890250 2.188890250
2.188890250 -0.000000000 2.188890250
ATOMIC_POSITIONS (crystal)
Si 0.000000000 -0.000000000 0.000000000
C 0.250000000 0.250000000 0.250000000
End final coordinates
即为弛豫后的晶格结构,可以看出晶格常数理论值为4.378Å。结构弛豫通过力的计算得到总能-位形曲面上的局部极小值,为了模拟实际,需要初始结构与实际情况足够接近。这里输出的density与ATOMIC_SPECIES中设置的原子质量有关。vc-relax计算包括多个自洽计算,查看自洽计算得到的总能用
grep '!' vc.out
输出为:
! total energy = -22.67776498 Ry
! total energy = -22.67819319 Ry
! total energy = -22.67819977 Ry
! total energy = -22.67819977 Ry
! total energy = -22.67821030 Ry
可见,这里vc-relax经历了5个自洽计算步骤后达到收敛,最后一个数值是结构弛豫计算得到的体系总能。其中,第5步是在第4步压强收敛之后,用第4步的结构重新做了一次scf计算,由于CELL的变化,平面波的个数随之变化,最后第5步计算的压强和第4步会有一定的差异,如果差异较大,则说明截断能未收敛,需要增加截断能,或用最后的结构重新启动vc-relax。
注意,自洽计算总能只具有相对意义,不同赝势计算得到的总能不能比较,用相同赝势计算的总能可以求差值。
1.2.1 自洽计算$\mathbf{k}$点的设置
对于周期性体系的平面波计算需要设置$\mathbf{k}$点网格。平面波方法中电子的电荷密度是布里渊区中的$\mathbf{k}$点网格求和得到的, $\rho(\mathbf{r})=\sum_{\mathbf{k} \in BZ} \omega_{\mathbf{k}} \lvert \psi_{\mathbf{k}}(\mathbf{r})\rvert ^{2}$, 其中,$\omega_{\mathbf{k}}$是依赖对称性的$\mathbf{k}$点权重。原则上更密的网格得到的电荷更准确,但是考虑到计算量,实际计算的$\mathbf{k}$点是较为有限的,设置
K_POINTS {automatic}
nk1, nk2, nk3, sk1, sk2, sk3
其中,前三个数nk1, nk2, nk3是倒格子三个基矢方向的$\mathbf{k}$点网格数,由于实际计算中,pw程序可能根据体系对称性约化$\mathbf{k}$点,因此k点个数有约化而不等于nk1*nk2*nk3。$nk_i$的值与第i个倒格子基矢长度成正比(对于满足正交的单元,$nk_i$的值与单元第i个基矢长度成反比),建议绝缘体设置$\mathbf{k}$点间距在$seperation=0.5Å^{-1}$以下。在QEtk中通过以下公式得到nk:$nk_i=max(1,round(2\pi \lvert \vec{b_i} \rvert /seperation)),i=1,2,3$,其中,$\lvert \vec{b_i} \rvert$是第i个倒格子矢量的长度,round(x)=floor(x+0.5)是四舍五入取整数。对于导体,nk的值需要大一些,需要同时结合degauss测试收敛;对于绝缘体,以上默认值足以得到相当准确的电荷密度。
后三个数sk1, sk2, sk3是设置$\mathbf{k}$点网格是否做平移,平移是相对于第i个基矢量方向的0点而言的。设置为0代表不做平移,即包括0点;设置为1代表做平移,不包括0点。
对于长度较大的单元基矢量,如分子、加入真空层的维度,以及其他采用超胞近似处理非周期性体系的维度,其中长度较大的维度对应的$\mathbf{k}$点网格只取坐标为0的点即可(相应nk等于1)。如果三个维度都是非周期性的,这时虽然QE平面波的计算仍然当作周期性体系来处理,对于$\mathbf{k}$点习惯上只选择$\Gamma$点。
K_POINTS {gamma}
1.2.2 占据数、展宽的设置
占据数和展宽是为了缓解对特别密的$\mathbf{k}$点网格的需求。对于导体,由于存在半满的能带,而只有占据的能级对于总能有贡献,费米能级附近的能级在自洽计算时可能出现相邻两次自洽迭代一次占据一次不占据,导致总能,尤其是单电子能级之和震荡的情况。计算总能时,除了使用足够密的$\mathbf{k}$点网格以外,设置占据数的展宽(smearing),即考虑费米能级附近degauss范围内的能级都对总能有一定贡献,可以让自洽计算在$\mathbf{k}$点不是特别密的时候即可收敛。degauss也不能取值过大,导体degauss的取值总在0.01Ry左右,过高的能级对总能的贡献是不符合实际情况的。对于绝缘体,不需要加入展宽。
建议设置occupation='smearing',smearing='gaussian',使用高斯展宽。高斯展宽对导体和绝缘体(含半导体,下同)是通用的,对于不同的材料调整degauss的值:(1)对于有带隙的材料,degauss=1d-9,也就是接近0就可以;(2)对于没有带隙的导体材料,degauss从小尝试到大,取值在0.01Ry量级,$\mathbf{k}$点足够多的基础上(见1.2.1),测试degauss一要总能几乎不变,二要输出中的“smearing contribution TS”足够小,建议小于$1\times10^{-4}$ Ry/atom。如果出现“smearing contribution TS”过大或者自洽计算超过nstep仍不能收敛,需要增加$\mathbf{k}$点并增减degauss,有必要时增加nbnd。
设置occupation='fixed'只适用于绝缘体,此时要增加nbnd的值以计算导带,默认只算到最高的价带,对于绝缘体,价带的个数是赝势文件中z_valence的值对CELL中的各个原子求和除以2(不含自旋轨道耦合)。
设置occupation='smearing', smearning='mv',mv(cold)、mp方法仅适用于导体。
虽然degauss这个变量有时称为electronic temperature,但是,在scf计算过程中,设置展宽的目的并不代表温度,费米-狄拉克分布虽然是自然的选择,但是其取值的长尾效应,需要较大的degauss,设置smearing=’fermi-dirac’会造成收敛相对较慢,一般不推荐这种设置。
$\mathbf{k}$点和展宽的设置,不仅对于总能,而且对于所有需要在整个布里渊区积分的物理量的计算收敛有影响,所以,$\mathbf{k}$点和展宽的选取,以所关心的物理量计算收敛为最终原则,同时考虑可接受的计算量。
1.2.3 截断能的设置
截断能ecutwfc和ecutrho的设置取决于赝势,一般赝势的作者会进行测试,测试包括结合能、能带、晶格常数、声子频率等。赝势的选择和使用见赝势教程。
1.3 自洽计算的输入文件
由于pw.x约定:在同一目录,并保持outdir、prefix一致时,先运行vc-relax、relax计算,在接着的scf、nscf、bands计算会读取之前弛豫后的结构,而忽略此时的结构设置。所以,此时做scf计算,修改的地方是:将calculation=’vc-relax’改成calculation=’scf’,其他部分与上一步输入文件相同。另外,vc-relax和relax计算最后一步包含了最终结构的scf计算,即vc-relax后面可以直接跟着bands计算,所以这里省去这一步骤。
1.4 能带计算的输入文件
能带计算,除了设置calculation=’bands’(’nscf’也可以),还需要设置$\mathbf{k}$点路径,我们参考文献[4]给出的特殊$\mathbf{k}$点坐标,选取了路径X-Γ-L。在relax计算的同一目录下,为了不和后处理bands.x的输入文件重名,将能带计算输入文件如下保存为nscf_b.inp,运行pw.x < nscf_b.inp > nscf_b.out。
&CONTROL
calculation='bands', disk_io='low', prefix='pwscf',
pseudo_dir='./', outdir='./tmp', verbosity='high'
tprnfor=.true., tstress=.true., forc_conv_thr=1.0d-5
/
&SYSTEM
ibrav= 0,
nat= 2, ntyp= 2,
occupations = 'smearing', smearing = 'gauss', degauss = 1.0d-9
ecutwfc= 50, ecutrho = 500,
/
&ELECTRONS
electron_maxstep = 100
conv_thr = 1.0d-9
mixing_mode = 'plain'
mixing_beta = 0.8d0
diagonalization = 'david'
/
&IONS
ion_dynamics='bfgs'
/
&CELL
press_conv_thr=0.1
/
ATOMIC_SPECIES
Si 1.0 Si.UPF
C 1.0 C.UPF
CELL_PARAMETERS (angstrom)
2.174000000 2.174000000 0.000000000
0.000000000 2.174000000 2.174000000
2.174000000 0.000000000 2.174000000
ATOMIC_POSITIONS (crystal)
Si 0.000000000 0.000000000 0.000000000
C 0.250000000 0.250000000 0.250000000
K_POINTS {crystal_b}
3
0.5 0.0 0.5 30
0.0 0.0 0.0 30
0.5 0.5 0.5 1
其中,K_POINTS部分采用了crystal_b,表示采用$\mathbf{k}$点的分数坐标能带模式,第一行代表计算3个特殊$\mathbf{k}$点之间的路径,下面3行是3个特殊$\mathbf{k}$点X-Γ-L的分数坐标以及权重,权重代表相邻特殊$\mathbf{k}$点之间计算的$\mathbf{k}$点数量(最后一个权重1不起作用,填入任意整数,但是,不可不填)。在crystal_b模式下,也可以用字母表示特殊$\mathbf{k}$点,具体定义见安装包(仅支持ibrav>0,13、14除外,可与坐标混用)/Doc/brillouin_zones.pdf。使用QE的后处理程序bands.x将能带结果转为易于画图的格式,将以下代码保存为bds.inp,运行bands.x < bands.inp > bands.out
&bands
prefix='pwscf',
outdir='tmp'
filband='bd.dat'
lp=.true.
/
这一个步骤是读取pw.x的输出,运行成功后,会生成filband所命名的文件,这里是bd.dat,bd.dat的内容见这里,是每个$\mathbf{k}$点的能级值,注意这里的能级数值只有相对的意义,能量的参考点是不确定的。各个$\mathbf{k}$点的能级值是从小到大排列的。
可见,bands.x只是后处理,pw.x输出中也能找到这些能级的值,另外tmp/pwscf.save/K00001/eigenval.xml等文件(每个K点一个文件夹)内也有足以画图的数据,且有效位数更高。bands.x的输入不支持windows换行符,需要结尾的“/”号之后额外换行,或用dos2unix处理之后在linux下编辑,windows版本的bands.exe也有这个问题。
通常,画能带图时,绝缘体用价带顶作为能量零点,导体用费米能级作为能量零点。接下来可以用QE自带的plotband.x画能带图,但是画图的自定义效果不够,这里不再介绍,这里为了用bd.dat画能带,使用python运行以下代码(或参考这里):
# -*- coding: utf-8 -*-
"""
@author: yyyu200@163.com
"""
import numpy as np
feig=open('bd.dat')
ymin=-20
ymax=20
nband=4 # this is the valence band number, for insulators only
dline=30 # vertical line intervals
l=feig.readline()
nbnd=int(l.split(',')[0].split('=')[1])
nks=int(l.split(',')[1].split('=')[1].split('/')[0])
eig=np.zeros((nks,nbnd),dtype=float)
for i in range(nks):
l=feig.readline()
count=0
if nbnd%10==0:
n=nbnd//10
else:
n=nbnd//10+1
for j in range(n):
l=feig.readline()
for k in range(len(l.split())):
eig[i][count]=l.split()[k]
count=count+1
feig.close()
import matplotlib as mpl
mpl.use('Agg')
import matplotlib.pyplot as plt
p1=plt.subplot(1, 1, 1)
F=plt.gcf()
F.set_size_inches([3,5])
lw=1.2 # line width
plt.xlim([0,nks-1]) # 201 points
plt.ylim([ymin,ymax])
#plt.xlabel(r'$k (\AA^{-1})$',fontsize=16)
plt.ylabel(r' E (eV) ',fontsize=16)
eig_vbm=max(eig[:,nband-1])
eig_cbm=min(eig[:,nband])
Gap=eig_cbm-eig_vbm
plt.title("Band gap="+str(Gap)[0:8]+" eV") # for insulators only
for i in range(nbnd):
line1=plt.plot( eig[:,i]-eig_vbm,color='r',linewidth=lw )
vline=dline
while vline<nks-1:
plt.axvline(x=vline, ymin=ymin, ymax=ymax,linewidth=lw,color='black')
vline=vline+dline
plt.xticks( (0,30,60), ('X', r'${\Gamma}$', 'L') )
plt.savefig('pwband.png',dpi=500)
计算能带结果与文献比较如下[5]。X-Γ带隙实验值为2.416eV,计算值为1.378eV,由于交换关联近似的原因,PBE计算出的带隙存在偏小的问题。在画能带图时,这里和大部分文献一样,能量的零点是取为价带顶,少数文献中取为禁带中点,说明清楚就可以,根据情况判断。
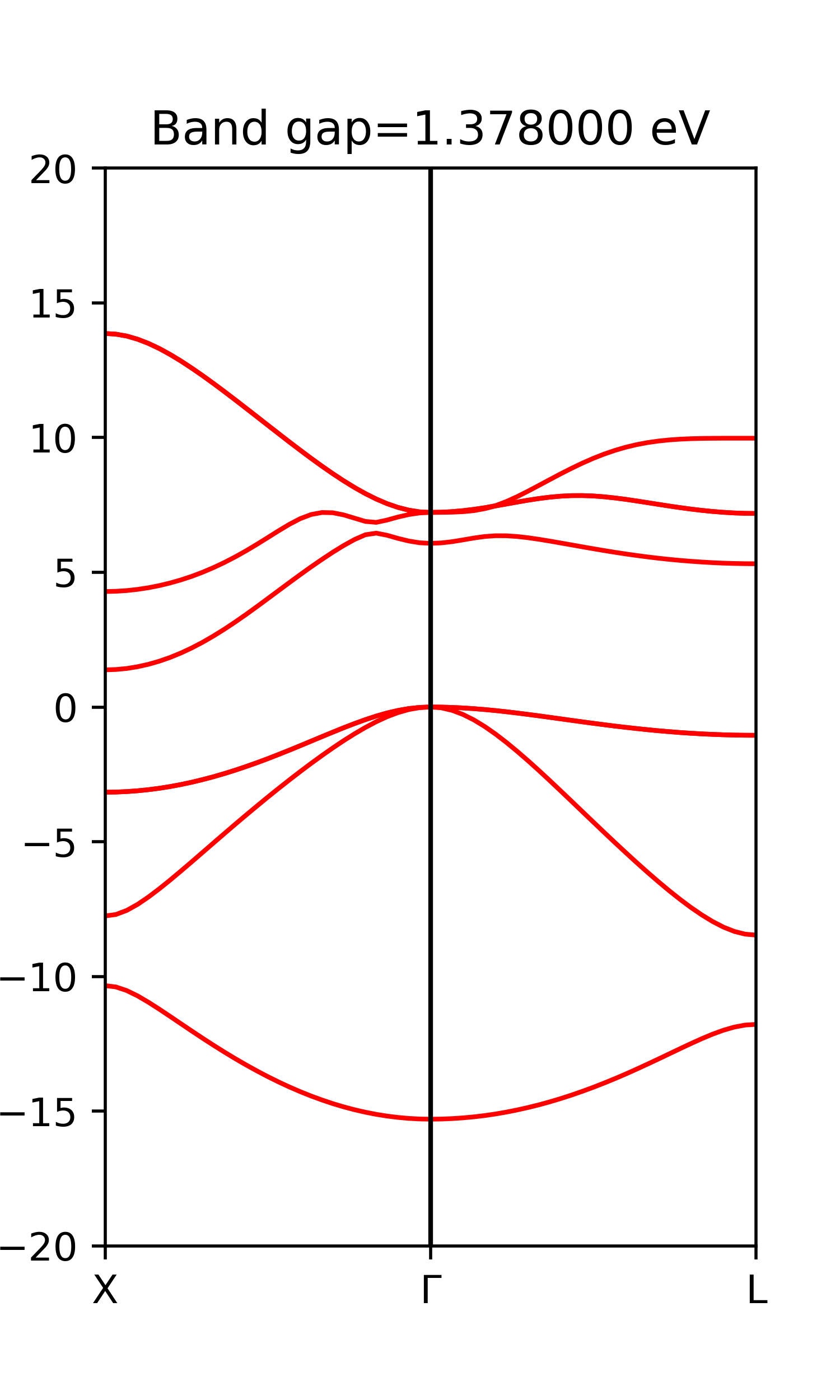
第一章计算文件及结果参考:这里。
2. 计算金属铜能带、功函数
导体的功函数是指导体的费米能级到真空能级的差值,对于绝缘体来说,一般用类似的概念,如电离势(ionic potential)和亲和能(electron affinity)来表示价带顶和导带底与真空能级的差值。pw.x通过scf计算得到自洽电荷密度,进而得到CELL内各个点的势能,同时得到体系的费米能级。在pw.x中,由于平面波程序的周期性边界条件,能级的参考点是不确定的,所以在块材(bulk)的计算中无法得到真空能级。为了同时得到费米能级和真空能级,需要建立平板(slab)模型,在CELL中留空,留空的区域即是真空,电荷密度基本为0,所以势能是常数,即真空能级。在slab计算中费米能级(绝缘体的VBM)由于表面态的存在而不准确,而slab内的平均势能和真空平均势能是准确的;在bulk计算中,费米能级和平均势能的差值是准确的。认为slab内平均势能和bulk平均势能是对应的,则可以得到真空能级和费米能级的差值,即功函数,参考文献[6]。
功函数与晶向、重构、吸附等密切相关,这里考虑金属Cu未重构的(110)表面,对于绝缘体的电离势可以通过类似方法得到。
2.1 计算铜的晶格常数、能带和态密度
用首先计算面心立方铜的原胞,用calculation='vc-relax'优化晶格常数。实验晶格常数为3.61Å。注意这里的smearing=’mp’(以及’mv’)只适用于导体。
&CONTROL
calculation='vc-relax', disk_io='low', prefix='pwscf',
pseudo_dir='./', outdir='./tmp', verbosity='high'
tprnfor=.true., tstress=.true., forc_conv_thr=1.0d-5
/
&SYSTEM
ibrav= 0,
nat= 1, ntyp= 1,
occupations = 'smearing', smearing = 'mp', degauss = 1.0d-2
ecutwfc= 50, ecutrho = 500,
/
&ELECTRONS
electron_maxstep = 100
conv_thr = 1.0d-9
mixing_mode = 'plain'
mixing_beta = 0.8d0
diagonalization = 'david'
/
&IONS
ion_dynamics='bfgs'
/
&CELL
press_conv_thr=0.1
/
ATOMIC_SPECIES
Cu 63.546 Cu.UPF
CELL_PARAMETERS (angstrom)
1.8050000 1.8050000 0.0000000
0.0000000 1.8050000 1.8050000
1.8050000 0.0000000 1.8050000
ATOMIC_POSITIONS (crystal)
Cu 0.000000000 0.000000000 0.000000000
K_POINTS {automatic}
13 13 13 0 0 0
运行awk '/Begin final coordinates/,/End final coordinates/{print $0}' vc.out得到:
Begin final coordinates
new unit-cell volume = 80.23493 a.u.^3 ( 11.88959 Ang^3 )
density = 8.87504 g/cm^3
CELL_PARAMETERS (angstrom)
1.811530376 1.811530376 0.000000000
0.000000000 1.811530376 1.811530376
1.811530376 0.000000000 1.811530376
ATOMIC_POSITIONS (crystal)
Cu 0.000000000 0.000000000 0.000000000
End final coordinates
可见,用PBE交换关联近似计算得到晶格常数理论值是1.811530376*2=3.6231Å。运行grep 'smearing contrib.' vc.out,
smearing contrib. (-TS) = 0.00000236 Ry
smearing contrib. (-TS) = 0.00000333 Ry
smearing contrib. (-TS) = 0.00000349 Ry
smearing contrib. (-TS) = 0.00000350 Ry
smearing contrib. (-TS) = 0.00000351 Ry
结果在可接受范围。
运行grep Fermi vc.out |tail -1,得到费米能级(以scf输出的费米能级为准)。
the Fermi energy is 12.7631 ev
计算态密度(这一步非计算功函数所需),nscf.inp输入如下,使用了$53^{3}=148877$个$\mathbf{k}$点的网格(对称性约化,实际计算了3654个点),以得到较为平滑的DOS曲线。
&CONTROL
calculation='nscf', disk_io='low', prefix='pwscf',
pseudo_dir='./', outdir='./tmp', verbosity='high'
tprnfor=.true., tstress=.true., forc_conv_thr=1.0d-5
/
&SYSTEM
ibrav= 0,
nat= 1, ntyp= 1,
occupations = 'smearing', smearing = 'mp', degauss = 1.0d-2
ecutwfc= 50, ecutrho = 500,
/
&ELECTRONS
electron_maxstep = 100
conv_thr = 1.0d-9
mixing_mode = 'plain'
mixing_beta = 0.8d0
diagonalization = 'david'
/
&IONS
ion_dynamics='bfgs'
/
&CELL
press_conv_thr=0.1
/
ATOMIC_SPECIES
Cu 63.546 Cu.UPF
CELL_PARAMETERS (angstrom)
1.8050000 1.8050000 0.0000000
1.8050000 0.0000000 1.8050000
0.0000000 1.8050000 1.8050000
ATOMIC_POSITIONS (crystal)
Cu 0.000000000 0.000000000 0.000000000
K_POINTS {automatic}
53 53 53 0 0 0
将以下输入参数保存为dos.inp,运行dos.x < dos.inp > dos.out
&dos
prefix='pwscf'
outdir='./tmp'
ngauss=1
degauss=1.5d-2
DeltaE=1.0d-2
fildos='cu.dos'
/
将nscf计算输入中K_POINTS改为如下,设置calculation='bands',计算能带。
K_POINTS {crystal_b}
6
0.5 0.5 0.5 50
0.0 0.0 0.0 50
0.5 0.0 0.5 20
0.625 0.25 0.625 0
0.375 0.375 0.75 50
0.0 0.0 0.0 1
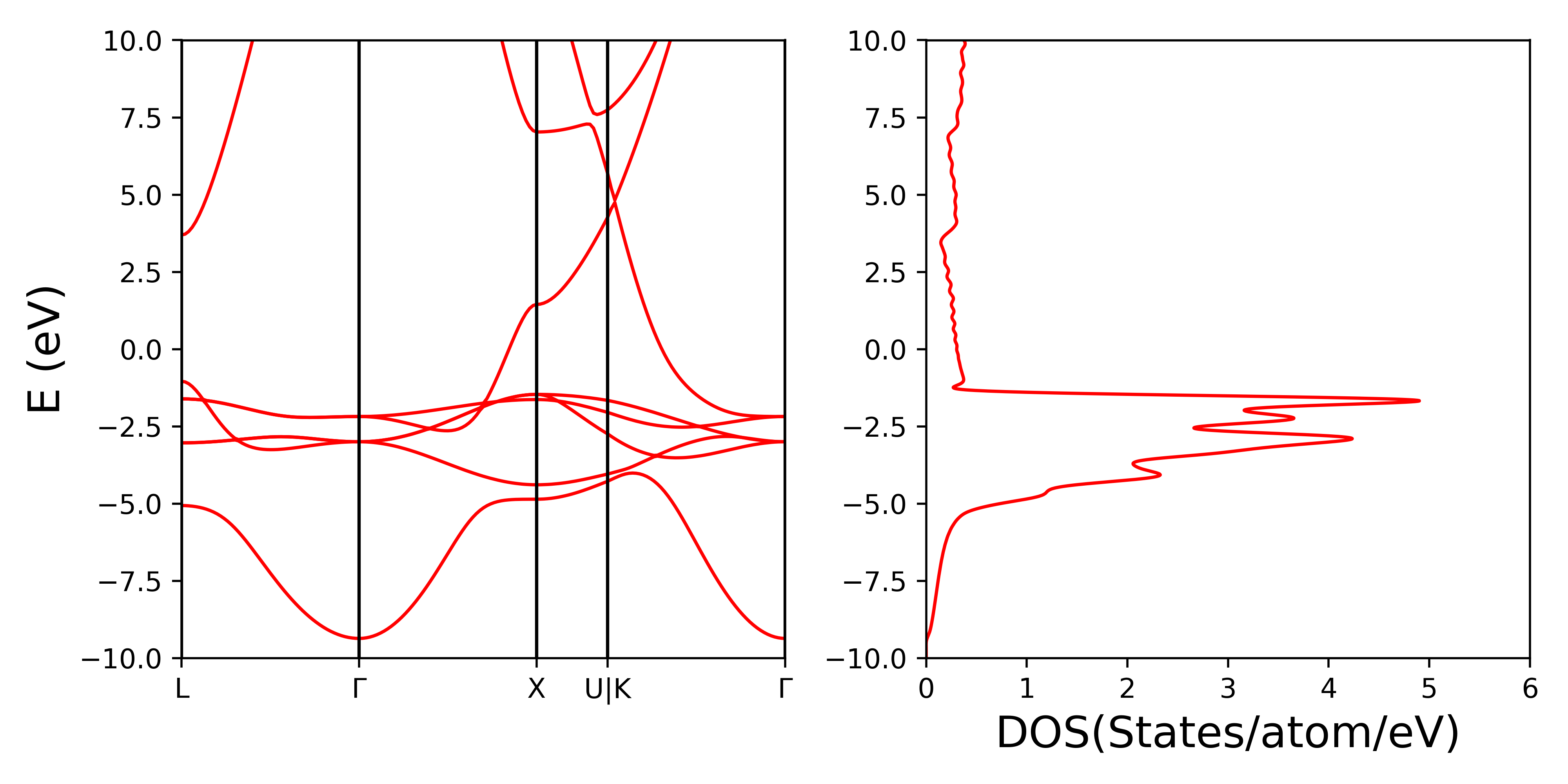
计算得到的能带和态密度如下图,画图脚本见这里,画图时要删掉第120个$\mathbf{k}$点(U点)的能级(能带和态密度不属于计算功函数的步骤,这里作为计算金属bulk的示例)。
2.2 建立slab模型并做结构驰豫
使用QEtoolkit中的SlabMaker工具建立slab模型,原理见建模教程。建立slab模型使用前一节得到的理论晶格常数,已知原胞时可以用QEtoolkit中的Prim2Unit工具,对原胞做变换\(P_{1}\)得到晶胞,
\(P_{1}=\quad \begin{pmatrix} -1 & 1 & 1 \\ 1 &-1 & 1 \\ 1 & 1 &-1 \\ \end{pmatrix} \quad\),
已知变换矩阵也可以用QEtoolkit中的Cell2Cell工具进行操作。
晶胞的POSCAR如下:
Cu unit cell
1.00000000000000
3.6231000000000000 0.0000000000000000 0.0000000000000000
0.0000000000000000 3.6231000000000000 0.0000000000000000
0.0000000000000000 0.0000000000000000 3.6231000000000000
Cu
4
Direct
0.0000000000000000 0.0000000000000000 0.0000000000000000
0.0000000000000000 0.5000000000000000 0.5000000000000000
0.5000000000000000 0.0000000000000000 0.5000000000000000
0.5000000000000000 0.5000000000000000 0.0000000000000000
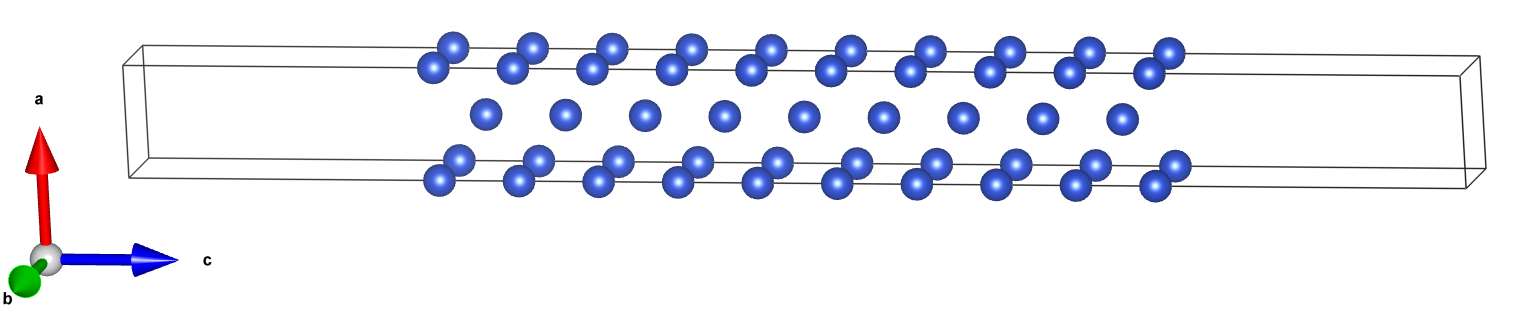
将晶胞的POCSAR(或cif,pw.x-input)上传至SlabMaker,填写密勒指数h=1,k=1,l=0,真空厚度vacuum=20,层数nlayer=5,z方向截取的起始面平移量shiftz=0,顶部删除原子数n_pop=1。
其中,n_pop是指删掉最顶端的一个原子(slab.pop()),得到的slab原子数是奇数,具有中心反演对称性,以保持slab的两个表面的对称性,消除两个表面不对称形成的偶极场(对于Cu的110面这个例子,由于对z方向做镜面加平移可以复原,不删除这个原子两个表面也是等势能的),z方向增加20Å真空层。得到pw.x的输入内容。
得到输入文件如下,这里的结构驰豫应该用calculation='relax'进行。在早期的研究中,常固定slab内部原子,只允许最接近表面的1~2层原子驰豫。现在常固定中间一个原子,等效于所有原子允许驰豫,可以避免slab的整体漂移(在relax中固定原子位置采用在原子坐标后面写“0 0 0”,分别固定坐标的三个分量)。
&CONTROL
title='Elemental:slab(20.0):(Cu)19:Orthorhombic P:Pmmm (47):primitive',
calculation='relax', pseudo_dir='./', outdir='./tmp', verbosity='high',
tprnfor=.true., tstress=.true., forc_conv_thr=1.0d-4, nstep=100,
/
&SYSTEM
ibrav= 0, nat= 19, ntyp= 1,
occupations = 'smearing', smearing = 'gauss', degauss = 1.0d-2,
ecutwfc = 50, ecutrho = 500,
/
&ELECTRONS
conv_thr = 1.0d-8
mixing_beta = 0.7d0
/
&IONS
/
&CELL
/
ATOMIC_SPECIES
Cu 63.546 Cu.UPF
CELL_PARAMETERS (angstrom)
2.5619185789 0.0000000000 0.0000000000
0.0000000000 3.6231000000 0.0000000000
0.0000000000 0.0000000000 43.0572672103
ATOMIC_POSITIONS (crystal)
Cu 0.0000000000 0.0000000000 0.2322488316
Cu 0.5000000000 0.5000000000 0.2619989614
Cu 0.0000000000 0.0000000000 0.2917490912
Cu 0.5000000000 0.5000000000 0.3214992210
Cu 0.0000000000 0.0000000000 0.3512493509
Cu 0.5000000000 0.5000000000 0.3809994807
Cu 0.0000000000 0.0000000000 0.4107496105
Cu 0.5000000000 0.5000000000 0.4404997403
Cu 0.0000000000 0.0000000000 0.4702498702
Cu 0.5000000000 0.5000000000 0.5000000000 0 0 0
Cu 0.0000000000 0.0000000000 0.5297501298
Cu 0.5000000000 0.5000000000 0.5595002597
Cu 0.0000000000 0.0000000000 0.5892503895
Cu 0.5000000000 0.5000000000 0.6190005193
Cu 0.0000000000 0.0000000000 0.6487506491
Cu 0.5000000000 0.5000000000 0.6785007790
Cu 0.0000000000 0.0000000000 0.7082509088
Cu 0.5000000000 0.5000000000 0.7380010386
Cu 0.0000000000 0.0000000000 0.7677511684
K_POINTS {automatic}
9 7 1 0 0 0
2.3 获取静电势并得到功函数
用pp.x得到静电势,如下文件保存为pp.inp,运行pp.x < pp.inp > pp.out,
&INPUTPP
outdir='./tmp'
plot_num=11
filplot='pp11.dat'
/
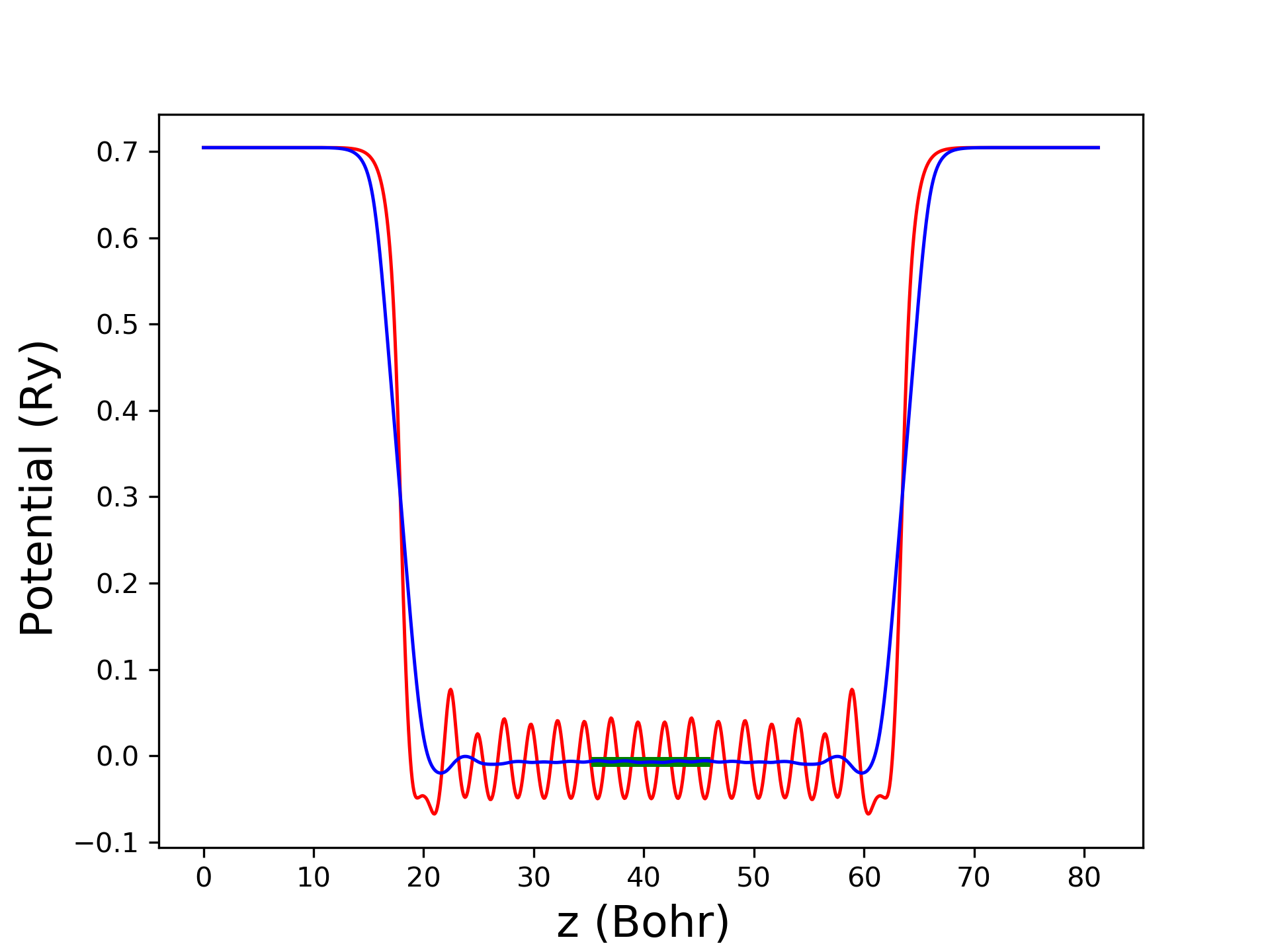
用average.x对面内势能取平均,并对垂直表面方向势能取移动平均值,average.x输入说明见这里,输入文件average.inp如下,运行average.x< average.inp >average.out:
1
pp11.dat
1
1000
3
4.8681
其中,最后一行4.8681是移动平均的窗口长度,设置为驰豫后slab中间部分的单元长度,单位是Bohr。输出avg.dat见这里,包括三列,第一列是z方向格点,第二列是势能在xy面内的平均,第三列是第二列在z方向移动平均。通过进一步拟合,得到slab中间势能为-0.00714Ry,真空势能为0.7042Ry。对于原胞的平均静电势能,可以对势能直接求平均值,得到原胞的平均势能是0.52914Ry,由前面计算得到原胞费米能级12.7631eV=0.9381Ry。
slab中费米能级是5.2158eV,不加原胞修正的功函数是4.369eV。
而加了原胞修正的功函数为:
\(0.7042-0.9381+[0.5294-(-0.0071)]=0.3026Ry=4.115eV\).
势能作图如下,红色线是xy面内平均势能,蓝色线是z方向移动平均,绿色线是slab内部平均势能示意。
2.4 加电场的自洽计算
对于slab模型、分子等,有时由于对称性的破坏,在平面波程序的周期性边界条件下,会造成人为的电场,需要加入一个dipole去消除;另外,有时需要研究体系在电场下的电荷变化,这时需要加电场的计算。在真空层中加入一个电偶极子,也就是引入一个沿着z方向(z是垂直表面方向,由edir设置)的锯齿形势能,这个势能在slab处上升,上升的梯度为电场强度eamp,在真空一个很小的宽度(eopreg)内下降。计算是否要做relax,根据实际情况定。设置需要用到edir,emaxpos,eopreg,eamp,tefield和dipfield。当slab位于Cell中间(约0.5)时(输入其余部分略):
&CONTROL
tefield=true., dipfield=.true.
/
&SYSTEM
edir = 3,
eamp = 0.001,
emaxpos=0.99,
eopreg=0.01,
/
偶极矩的位置在z方向的0.99~1.0之间。
对于slab,总是有两个面,而这两个面不相同时,周期性边界条件会造成一个额外的电场,有时需要加偶极修正,这时设置eamp=0,偶极电场大小通过自洽计算自动生成,修正之后真空部分的势能应该是不随z坐标明显变化的。最近新增的assume_isolated='2D'也能实现类似的功能,参考相关文献。
注:QE中另一种加电场的方法是用berry phase(BP)加入电场,适用的场合不同,BP方法要求体系是没有真空层的,或者有真空层但是电场在slab的面内(x,y方向)。berry phase电场设置参数
lelfield=.true., nberrycyc=3
...
efield_cart(3)=0.001d0
对于导体不适用加电场,(1)dipole方法,金属slab加电场会让正负电荷在两个表面积累,这种电荷转移的量很大,QE计算几乎不可能收敛。(2)berry phase方法,由于berry phase是在倒空间一个闭合的曲面上的积分,金属的占据态对应$\mathbf{k}$点集合不是闭合的曲面,无法定义Berry phase。
2.5 静态及光学介电常数计算
在2.4节的基础上,可以计算固体的静态及光学介电常数,参考安装包中的q-e-qe-6.5\PW\examples\example10,原理和参考文献看一下其中的README,分别做两次相反方向小电场的自洽计算,得到极化的改变量,从而得到介电常数。
计算静态介电常数,只要在上面光学介电常数的基础上,允许结构做relax就可以得到。
在example10中,使用的是berry phase电场,对于含有真空的cell,则可以用dipole电场。两种加电场方法都可以得到比较准确的结果。需要注意的是相关物理量(总能、力)的收敛问题,相关阈值要适当调小,经过测试,即使非常接近铁电的体系仍然可以得到合理的结果。
3. 计算氮化硼的光学性质
计算闪锌矿结构立方氮化硼的介电函数。计算固体光学性质的方法,比较先进的是通过GW-BSE方程加入电子的自能(GW)、电子-空穴相互作用修正(BSE),以及含时密度泛函理论(TDDFT)等方法[17],在QE中有TDDFPT,GWL,Yambo等模块发展相关的方法。
TDDFT与GW-BSE计算相比,GW-BSE计算使用格林函数,直接构造了物理过程,是计算光学性质的标准方法;TDDFT基于电子密度,包含了各种作用但并不那么直观,重要的是TDDFT计算量比GW-BSE小很多。这两种方法是互补的关系,原则上都是精确的。TDDFT使用的交换关联泛函十分关键,原则上需要非局域泛函,如Hybrid functional、meta GGA、OEP等(后两种暂未实现,hybrid只支持turbo_lanczos.x)。
3.1 用epsilon.x计算光学性质
这里,首先介绍一种基于LDA/GGA的通过Kohn-Sham轨道跃迁矩阵元计算介电常数的QE后处理程序epsilon.x。编译了QE的PP模块(make pp)之后,目录qe/PP/src下会生成这个可执行文件。epsilon.x计算能级间偶极跃迁得到介电常数,根据以下公式得到介电常数虚部[7]:
$\epsilon_2 (\omega)= \frac{4 \pi ^2} {m^2\omega^2} \sum_{V,C} {\int_{BZ} {d^3 k \frac {2}{(2\pi)^{3}} \lvert eM_{CV}(K) \rvert ^2 \cdot \delta[E_{C}(k)-E_{V}(k)-h\omega]}}$
而介电常数实部可以由虚部通过Kramers-Kronig关系变换得到。通过能带结构,可以计算得到全部的光学常数。
epsilon.x计算目前仅支持模守恒赝势。首先优化晶格常数,运行pw.x < vc.inp > vc.out,vc.inp如下:
&CONTROL
calculation='vc-relax', disk_io='low', prefix='pwscf',
pseudo_dir='./', outdir='./tmp', verbosity='high'
tprnfor=.true., tstress=.true., forc_conv_thr=1.0d-5
/
&SYSTEM
ibrav= 0,
nat= 2, ntyp= 2,
occupations = 'fixed'
ecutwfc= 80, ecutrho = 320,
/
&ELECTRONS
electron_maxstep = 100
conv_thr = 1.0d-9
mixing_mode = 'plain'
mixing_beta = 0.8d0
diagonalization = 'david'
/
&IONS
ion_dynamics='bfgs'
/
&CELL
press_conv_thr=0.1
/
ATOMIC_SPECIES
B 10.811 B_ONCV_PBE-1.0.upf
N 14.00674 N.oncvpsp.upf
CELL_PARAMETERS (angstrom)
1.807500000 1.807500000 -0.000000000
0.000000000 1.807500000 1.807500000
1.807500000 -0.000000000 1.807500000
ATOMIC_POSITIONS (crystal)
B 0.000000000 0.000000000 0.000000000
N 0.250000000 0.250000000 0.250000000
K_POINTS {automatic}
13 13 13 0 0 0
赝势文件见 B_ONCV_PBE-1.0.upf N.oncvpsp.upf。
得到晶格常数PBE计算值3.6215Å。光学性质需要较密的$\mathbf{k}$点,并且需要关掉对称性,设置nosym=.true,noinv=.true,计算的带个数nbnd要大一些,做一次nscf计算:
&CONTROL
calculation='nscf', disk_io='low', prefix='pwscf',
pseudo_dir='./', outdir='./tmp', verbosity='high'
tprnfor=.true., tstress=.true., forc_conv_thr=1.0d-5
/
&SYSTEM
ibrav= 0,
nat= 2, ntyp= 2,
occupations = 'smearing', smearing='gauss', degauss=1d-9,
ecutwfc= 80, ecutrho = 320,
nbnd=30
nosym=.true,
noinv=.true,
/
&ELECTRONS
electron_maxstep = 100
conv_thr = 1.0d-9
mixing_mode = 'plain'
mixing_beta = 0.8d0
diagonalization = 'david'
/
&IONS
ion_dynamics='bfgs'
/
&CELL
press_conv_thr=0.1
/
ATOMIC_SPECIES
B 10.811 B_ONCV_PBE-1.0.upf
N 14.00674 N.oncvpsp.upf
CELL_PARAMETERS (angstrom)
1.810733563 1.810733563 0.000000000
-0.000000000 1.810733563 1.810733563
1.810733563 -0.000000000 1.810733563
ATOMIC_POSITIONS (crystal)
B 0.000000000 0.000000000 0.000000000
N 0.250000000 0.250000000 0.250000000
K_POINTS {automatic}
13 13 13 0 0 0
以下内容保存为epsilon.inp,运行epsilon.x < epsilon.inp > epsilon.out:
&inputpp
outdir='./tmp'
calculation='eps'
/
&energy_grid
smeartype='gauss'
intersmear=0.50
intrasmear=0.0
wmin=0.0
wmax=60.0
nbndmin=1
nbndmax=0
nw=2000
shift=0.0
/
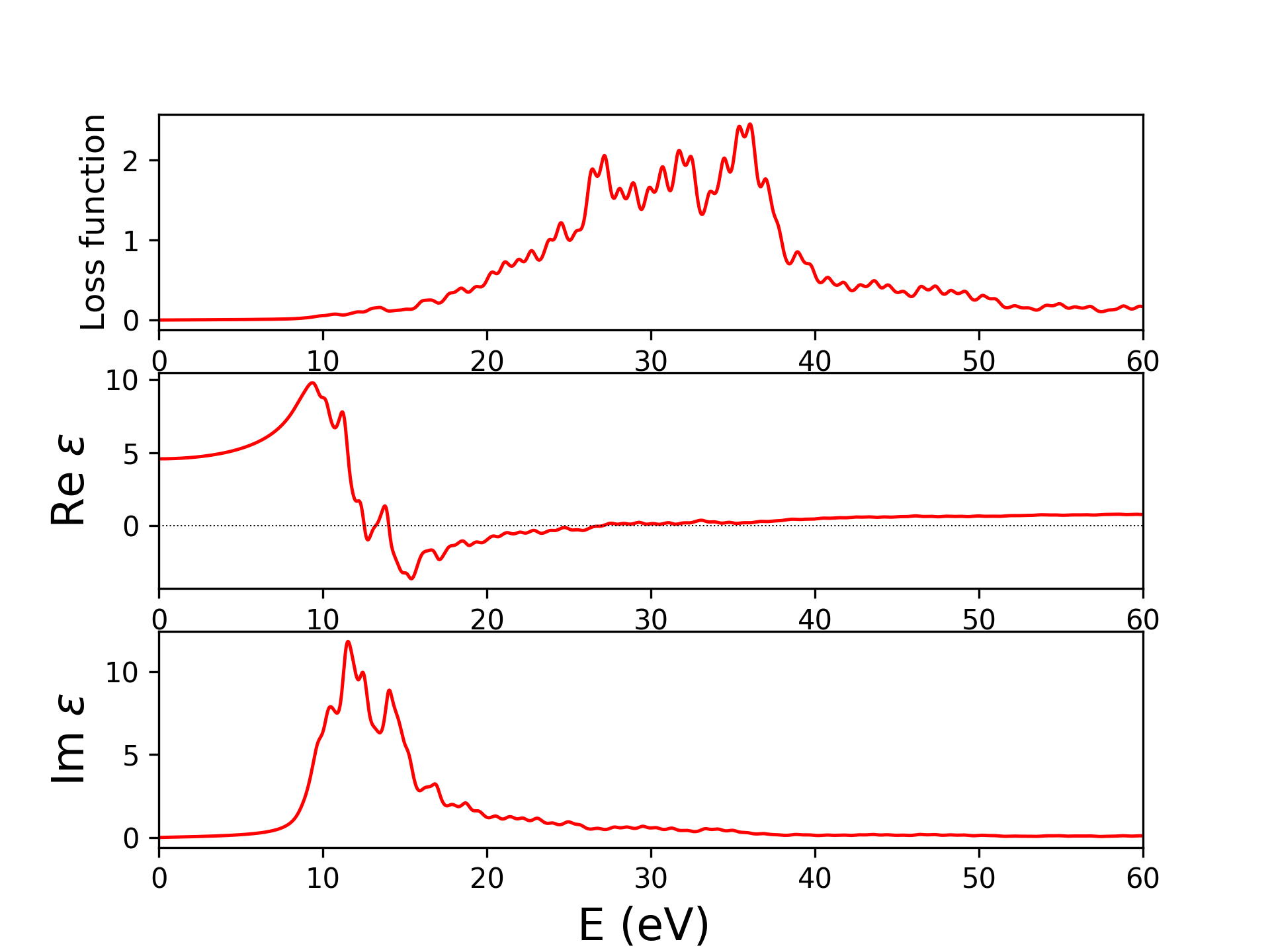
输出4个文件eels_pwscf.dat,epsi_pwscf.dat,epsr_pwscf.dat,ieps_pwscf.dat,分别是eels谱(电子能量损失谱),介电函数的虚部、实部,虚轴上的介电函数。前三个结果见下图,与实验及其他计算结果比较见[8]。
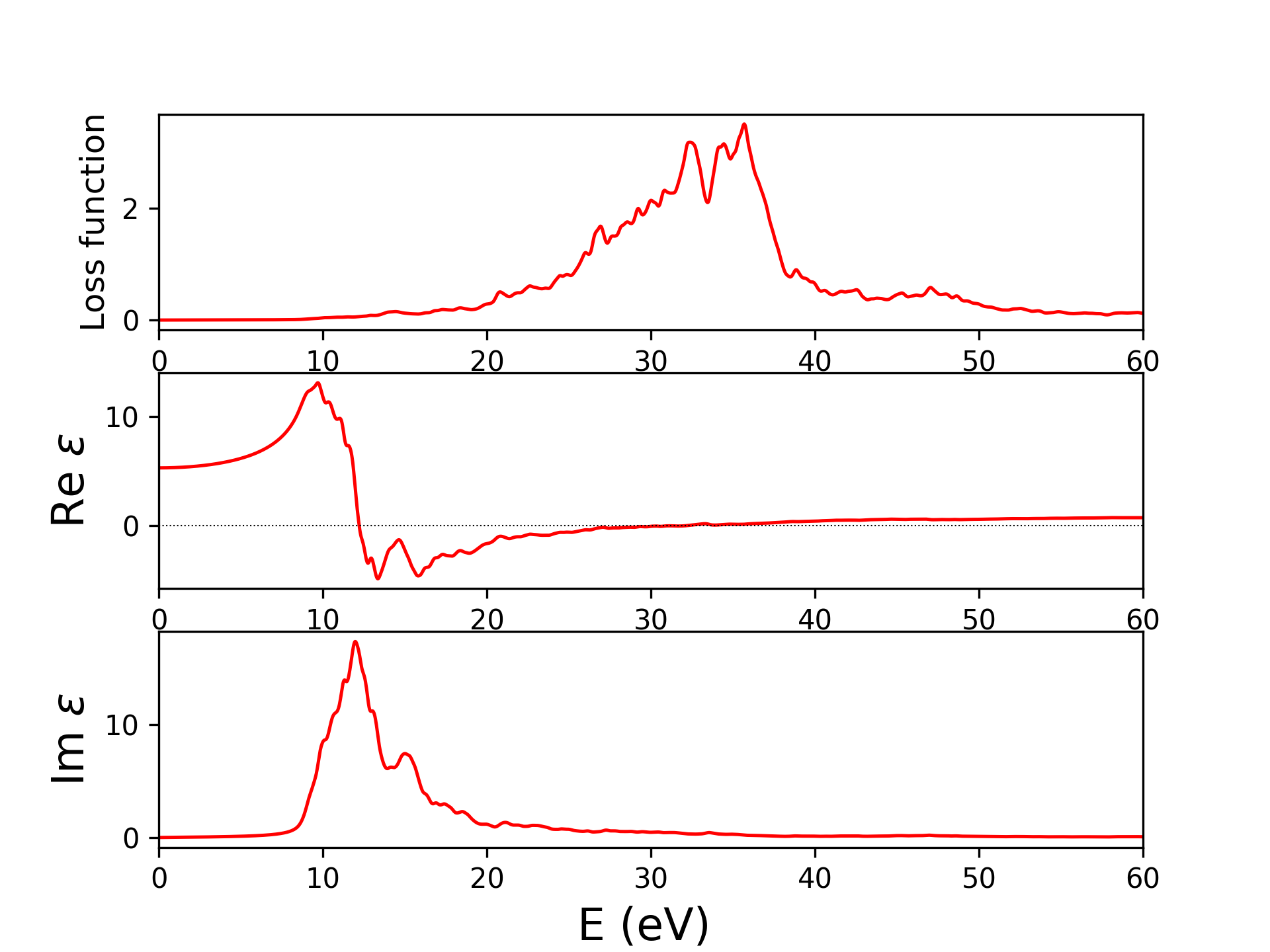
固体介电常数、电导率、折射率一般来说都是复数,知道其一可以确定其余两个,进而还可以得到吸收系数,反射率,三者关系如下表[9]:

吸收系数 $\alpha= \frac{2k \omega }{c}$
反射率 $R=\frac{(1-n)^2+k^2}{(1+n)^2+k^2}$
3.2 用tddfpt模块计算光学性质
TDDFPT模块[15],包括适用于分子体系的turbo_lanczos.x(以及turbo_davidson.x),和计算固体的turbo_eels.x,以及它们的后处理程序turbo_spectrum.x。turbo_lanczos.x只支持单个$\mathbf{k}$点($\Gamma$点),对于固体要用超胞近似。下面使用turbo_eels.x模块,turbo_eels.x需要指定转移动量,一般地,介电函数是动量和频率的函数,这在电子能量损失谱(EELS)中较容易理解,介电函数动量谱的意义是体系在空间非均匀电场下的响应,EELS实验上是入射、出射电子的动量变化,光学性质是动量趋向于0的极限。
同3.1节的BN材料,首先进行一次scf计算。
&CONTROL
calculation='scf', disk_io='low', prefix='pwscf',
pseudo_dir='./', outdir='./tmp', verbosity='high'
tprnfor=.true., tstress=.true., forc_conv_thr=1.0d-5
/
&SYSTEM
ibrav= 0,
celldm(1) = 1.8897261328856432, ! a.u. to Angst
nat= 2, ntyp= 2,
occupations = 'smearing', smearing='gauss', degauss=1d-9,
!occupations = 'fixed',
ecutwfc= 80, ecutrho = 320,
nbnd=30,
/
&ELECTRONS
electron_maxstep = 100
conv_thr = 1.0d-9
mixing_mode = 'plain'
mixing_beta = 0.8d0
diagonalization = 'david'
/
&IONS
ion_dynamics='bfgs'
/
&CELL
press_conv_thr=0.1
/
ATOMIC_SPECIES
B 10.811 B_ONCV_PBE-1.0.upf
N 14.00674 N.oncvpsp.upf
CELL_PARAMETERS (alat= 1.88972613)
1.810733563 1.810733563 0.000000000
-0.000000000 1.810733563 1.810733563
1.810733563 -0.000000000 1.810733563
ATOMIC_POSITIONS (crystal)
B 0.000000000 0.000000000 0.000000000
N 0.250000000 0.250000000 0.250000000
K_POINTS {automatic}
8 8 8 0 0 0
turbo_eels.x的输入如下,需要指定一个接近0的动量q值[16]。itermax是时间步数。
&lr_input
prefix='pwscf',
outdir='./tmp'
restart_step=10,
restart=.false.
/
&lr_control
itermax = 5000,
ipol=1,
q1 = 0.001d0,
q2 = 0.0d0,
q3 = 0.0d0,
/
运行turbo_spectrum.x,输入如下,注意使用了插值extrapolation,展宽值epsil选取对结果影响较大。
&lr_input
prefix='pwscf',
outdir='./tmp'
eels = .true.
itermax0 = 5000
itermax = 10000
extrapolation="osc"
epsil=0.0350
units=1
start=0.0d0
increment=0.001d0
end=60d0
/
输出pwscf.plot_eps.dat,画图如下。
4. 声子谱的计算
计算GaAs的声子谱。QE中声子计算是通过DFPT进行的,通过加入berry phase电场计算电荷响应得到介电常数,新版本(>=6.x)也开始支持finite displacements method。参考安装包q-e-qe-6.3\PHonon\examples\ example1 ($\Gamma$点)和example2 (色散),画图这里用的是python。PHonon模块在同一目录重新计算时需要删除部分旧文件(./tmp/_ph0),否则会报错退出。
首先优化晶格常数,用pw.x进行一次vc-relax计算(晶格常数实验值5.715Å,计算值5.585Å)。GGA计算出的极化往往较实际偏大,而LDA往往偏小,在极化$4\pi P$接近电位移矢量D时介电常数发散($4\pi P$总是小于D),$\epsilon=1+\frac{4\pi P}{D-4\pi P}$,所以,极化偏大对介电常数影响较大。这里GaAs按照文献中的做法使用了LDA模守恒赝势,赝势文件见Ga,As。
&CONTROL
calculation='vc-relax', disk_io='low', prefix='pwscf',
pseudo_dir='./', outdir='./tmp', verbosity='high'
tprnfor=.true., tstress=.true., forc_conv_thr=1.0d-5, etot_conv_thr=1.0d-5
/
&SYSTEM
ibrav= 2,
celldm(1) = 10.7997848
nat= 2, ntyp= 2,
occupations = 'fixed', nbnd=10
ecutwfc= 120, ecutrho = 480,
input_dft='pz'
/
&ELECTRONS
electron_maxstep = 100, conv_thr = 1.0d-9, mixing_mode = 'plain'
mixing_beta = 0.7d0, diagonalization = 'david'
/
&IONS
ion_dynamics='bfgs'
/
&CELL
press = 0.00 ,
press_conv_thr=0.1
cell_dynamics = 'bfgs' ,
/
ATOMIC_SPECIES
Ga 69.7230 Ga.SG15.LDA.UPF
As 74.9216 As.SG15.LDA.UPF
ATOMIC_POSITIONS (crystal)
Ga 0.000000000 0.000000000 0.000000000 0 0 0
As 0.250000000 0.250000000 0.250000000
K_POINTS {automatic}
11 11 11 0 0 0
4.1 单个$\mathbf{q}$点的ph.x计算
vc-relax计算之后用最终结构做一次scf计算(否则后续计算会出错)。
将以下文件保存为ph.inp,运行ph.x < ph.inp > ph.out
phonons of GaAs at Gamma
&inputph
tr2_ph=1.0d-14,
prefix='pwscf',
outdir='./tmp',
epsil=.true.,
amass(1)=69.7230,
amass(2)=74.9216,
fildyn='gs.dynG',
/
0.0 0.0 0.0
fildyn是输出动力学矩阵文件,共有($\mathbf{q}$点数+1)个,后缀为0的文件是这些$\mathbf{q}$点的坐标,后缀为1至$\mathbf{q}$点数的文件是动力学矩阵、频率等值,在$\Gamma$点还有光学介电常数、有效电荷等值。在输出ph.out中可以找到高频介电常数张量$\epsilon_{ij}(\infty)$(实验值10.86,Ref.[10]),以及玻恩有效电荷张量$Z_{ij}$(实验值2.07):
Dielectric constant in cartesian axis
( 13.578406136 -0.000000000 0.000000000 )
( -0.000000000 13.578406136 -0.000000000 )
( 0.000000000 -0.000000000 13.578406136 )
Effective charges (d Force / dE) in cartesian axis
atom 1 Ga
Ex ( 2.04400 0.00000 0.00000 )
Ey ( 0.00000 2.04400 -0.00000 )
Ez ( 0.00000 -0.00000 2.04400 )
atom 2 As
Ex ( -2.04051 -0.00000 -0.00000 )
Ey ( -0.00000 -2.04051 0.00000 )
Ez ( -0.00000 0.00000 -2.04051 )
此时计算出$\Gamma$点声子频率:
Diagonalizing the dynamical matrix
q = ( 0.000000000 0.000000000 0.000000000 )
**************************************************************************
freq ( 1) = -2.183574 [THz] = -72.836200 [cm-1]
freq ( 2) = -2.183574 [THz] = -72.836200 [cm-1]
freq ( 3) = -2.183574 [THz] = -72.836200 [cm-1]
freq ( 4) = 7.777154 [THz] = 259.417942 [cm-1]
freq ( 5) = 7.777154 [THz] = 259.417942 [cm-1]
freq ( 6) = 7.777154 [THz] = 259.417942 [cm-1]
**************************************************************************
但是,可以看到ph.x输出的$\Gamma$点声学声子不等于0,且没有LO-TO分裂。在声子平衡位置,由对称性可知LO与TO是简并的。但是,极性材料中LO与长程库仑作用耦合而TO不耦合造成LO-TO分裂[11]。为了加入Acoustic Sum Rule和non-analytic term修正,将以下文件保存为dynmat.inp,运行dynmat.x < dynmat.inp > dynmat.out
&input
fildyn='gs.dynG'
q(1)=0,q(2)=0,q(3)=0.1
asr='crystal'
filout='dynmat.dat'
/
其中,q(1,2,3)是LO-TO分裂的方向,对于立方晶系各向同性不全等于0即可。dynmat.x加入了非解析项修正,对于q=0点加入了声学声子求和规则,再次对角化动力学矩阵。输出的dynmat.out中可以找到$\Gamma$点声子频率如下(实验值:TO=271$cm^{-1}$,LO=293$cm^{-1}$)。第三列是红外强度。
# mode [cm-1] [THz] IR
1 -0.00 -0.0000 0.0000
2 0.00 0.0000 0.0000
3 0.00 0.0000 0.0000
4 259.42 7.7771 2.6645
5 259.42 7.7771 2.6645
6 277.32 8.3139 2.6645
4.2 声子色散计算
声子色散是在倒空间特殊路径上声子频率的连线,这里倒空间和电子能带计算中的倒空间含义是一样的,但是,对于声子的波矢习惯上记作$\mathbf{q}$,电子波矢习惯上记作$\mathbf{k}$。
首先,用ph.x在$\mathbf{q}$点网格上通过DFPT计算动力学矩阵,输入ph.inp如下(需要在scf计算之后运行,如果接在vc-relax之后运行ph.x,运行q2r.x时会出错),运行ph.x < ph.inp > ph.out:
phonons of GaAs q-grid
&inputph
tr2_ph=1.0d-12,
prefix='pwscf',
outdir='./tmp',
amass(1)=69.7230,
amass(2)=74.9216,
fildyn='gs666.dyn',
ldisp=.true.,
nq1=6, nq2=6, nq3=6
/
ph.x计算结束后(这一步ph.x计算量很大,24核算了约30小时),q2r.x将动力学矩阵由q空间变换到实空间,输入q2r.inp如下,运行q2r.x < q2r.inp > q2r.out:
&input
fildyn='gs666.dyn', zasr='simple', flfrc='gs666.fc'
/
其中,flfrc是q2r.x输出的力常数文件。在输出文件q2r.out中需要有以下内容。
fft-check success (sum of imaginary terms < 10^-12)
matdyn.x通过实空间nq1×nq2×nq3超胞上interatomic force constants计算任意$\mathbf{q}$点声子频率。对于非立方材料各项异性的LO-TO分裂需要将$\Gamma$点写两次,立方材料则不需要。将以下保存为matdyn.inp,运行matdyn.x < matdyn.inp > matdyn.out。计算声子色散时,一般设置q_in_band_form=.true.,只给出每一段$\mathbf{q}$点路径的端点;另一种可行的设置,即使用默认的fasle,并给出每一个$\mathbf{q}$点坐标。$\mathbf{q}$点定义见安装包/Doc/brillouin_zones.pdf,可以用字母表示特殊$\mathbf{q}$点,也可以用笛卡尔坐标(2$\pi$/alat为单位)。有需要时可以设置q_in_cryst_coord=.true.,$\mathbf{q}$点为分数坐标;默认为false,表示直角坐标。
&input
asr='simple',
amass(1)=69.7230,
amass(2)=74.9216,
flfrc='gs666.fc',
flfrq='gs.freq',
q_in_band_form=.true.,
/
6
gG 40
X 10
U 0
K 40
gG 40
L 1
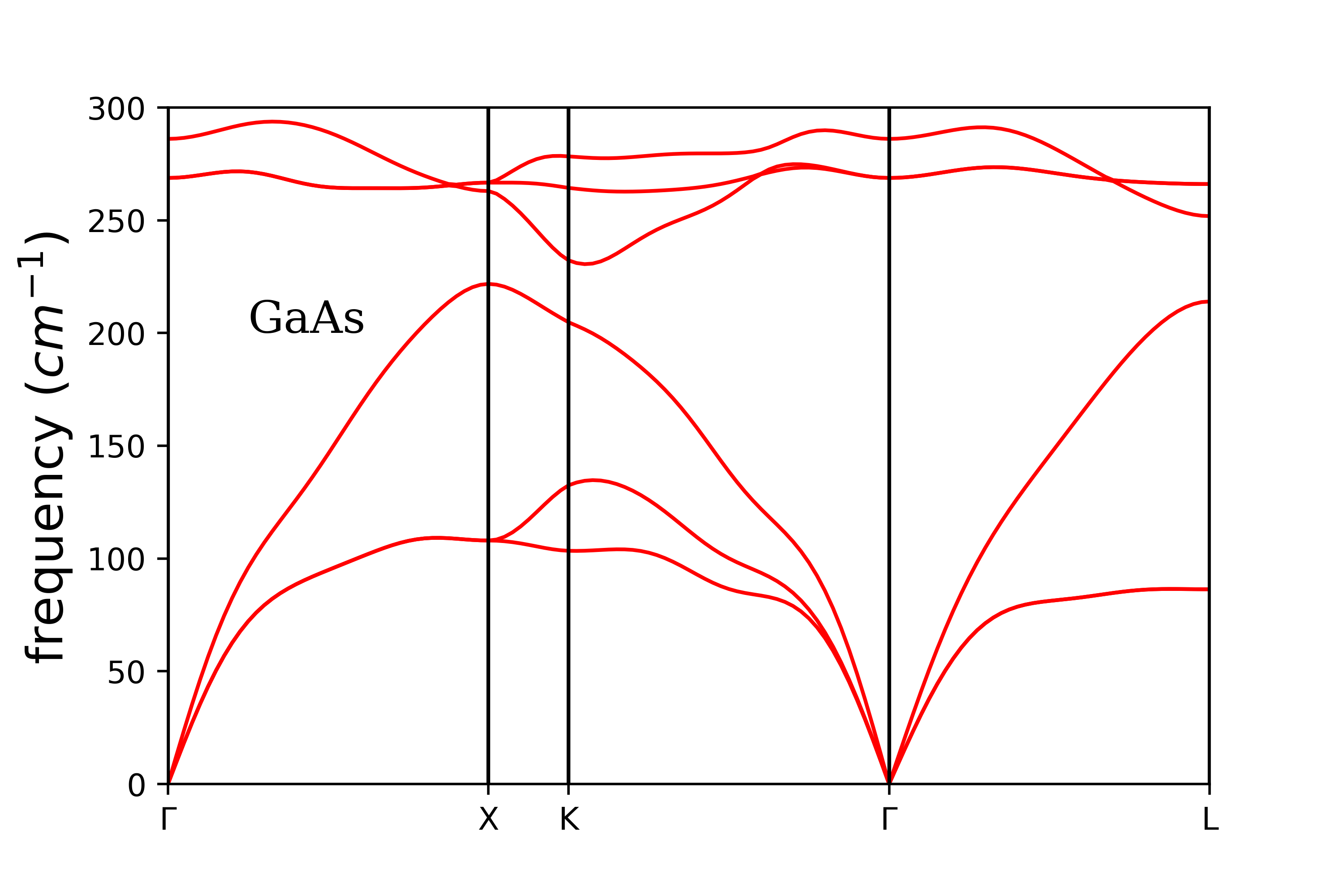
输出’gs.freq’文件格式和1.4节计算电子能带的bd.dat文件格式是一致的,画图参考1.4节的脚本,声子色散见下图,画图时要删掉第50个(U点)和第51个(K点)$\mathbf{q}$点中的一个,结果与文献中比较见[10]。matdyn.x也可以用来计算声子态密度、电声子耦合系数(见qe_release_6.4/PHonon/examples/example03)。
4.3 电子-声子耦合系数
4.3.1 使用PHonon模块计算电子-声子耦合系数
面心立方Pb,晶格常数9.316 bohr(4.93Å)。与4.2节同样的原因,使用LDA赝势,vc-relax得到优化晶格常数是9.2083 bohr。
以下计算分为7个步骤(第5步非必需),其中q2r.x,matdyn.x,lambda.x这三个模块运行时不用mpi并行。
(1) 较密$\mathbf{k}$点的scf计算
输入如下,注意这一步加入了la2F=.true.,$\mathbf{k}$点设置为36×36×36的网格,此处必须包含$\Gamma$点且是第2步scf计算$\mathbf{k}$点、声子ph.x计算$\mathbf{q}$点的整数倍。计算会输出一个./tmp/pwscf.a2Fsave文件,会在后面的ph.x计算中读取。
&CONTROL
calculation='scf', disk_io='low', prefix='pwscf',
pseudo_dir='./', outdir='./tmp', verbosity='high'
tprnfor=.true., tstress=.true., forc_conv_thr=1.0d-5
/
&SYSTEM
ibrav= 2,
celldm(1) = 9.2083, nat= 1, ntyp= 1,
nat= 1, ntyp= 1,
occupations = 'smearing', smearing = 'mp', degauss = 2.5d-2
ecutwfc= 50, ecutrho = 500,
la2F=.true.
/
&ELECTRONS
conv_thr = 1.0d-9,
mixing_beta = 0.7d0,
diagonalization = 'david'
/
&IONS
ion_dynamics='damp'
/
&CELL
press = 0.00 ,
press_conv_thr=0.1
cell_dynamics = 'bfgs' ,
/
ATOMIC_SPECIES
Pb 207.2 Pb.pz-dn-rrkjus_psl.1.0.0.UPF
ATOMIC_POSITIONS (crystal)
Pb 0.000000000 0.000000000 0.000000000
K_POINTS {automatic}
36 36 36 0 0 0
(2) 正常$\mathbf{k}$点的scf计算
这一步并非必须,是考虑计算量的取舍,接下来的ph.x计算中电子计算的$\mathbf{k}$点、截断、电荷密度是读取这一步的结果进行的,la2F不设置,不覆盖上一步的pwscf.a2Fsave输出。
&CONTROL
calculation='scf', disk_io='low', prefix='pwscf',
pseudo_dir='./', outdir='./tmp', verbosity='high'
tprnfor=.true., tstress=.true., forc_conv_thr=1.0d-5
/
&SYSTEM
ibrav= 2,
celldm(1) = 9.2083, nat= 1, ntyp= 1,
nat= 1, ntyp= 1,
occupations = 'smearing', smearing = 'mp', degauss = 2.5d-2
ecutwfc= 50, ecutrho = 500,
/
&ELECTRONS
conv_thr = 1.0d-9,
mixing_beta = 0.7d0,
diagonalization = 'david'
/
&IONS
ion_dynamics='damp'
/
&CELL
press = 0.00 ,
press_conv_thr=0.1
cell_dynamics = 'bfgs' ,
/
ATOMIC_SPECIES
Pb 207.2 Pb.pz-dn-rrkjus_psl.1.0.0.UPF
ATOMIC_POSITIONS (crystal)
Pb 0.000000000 0.000000000 0.000000000
K_POINTS {automatic}
18 18 18 0 0 0
(3) ph.x计算电声子系数
ph.inp文件中设置:
(a) qe-6.6/PHonon/PH/q_points.f90第85行做以下修改,重新编译PHonon模块。这时可以得到$\mathbf{q}$点网格坐标及权重,在后面的lambda.x计算会用到,
- write(stdout, '(5x,i3, 3f14.9)') iq, x_q(1,iq), x_q(2,iq), x_q(3,iq)
+ write(stdout, '(5x,i3, 4f14.9)') iq, x_q(1,iq), x_q(2,iq), x_q(3,iq), wq(iq)
此时ph.x输入如下:
Electron-phonon coefficients
&inputph
tr2_ph=1.0d-10,
prefix='pwscf',
fildvscf='pbdv',
amass(1)=207.2,
outdir='./tmp',
fildyn='pb.dyn',
electron_phonon='interpolated',
el_ph_sigma=0.005,
el_ph_nsigma=10,
trans=.true.,
ldisp=.true.
nq1=4, nq2=4, nq3=4
/
其中,el_ph_nsigma=10是测试10个不同的展宽值,以el_ph_sigma=0.005为间隔(同时也是初始值)。
重新编译ph.x的输出可以找到如下内容,包括了$\mathbf{q}$点的权重
Dynamical matrices for ( 4, 4, 4) uniform grid of q-points
( 8 q-points):
N xq(1) xq(2) xq(3)
1 0.000000000 0.000000000 0.000000000 0.015625000
2 -0.250000000 0.250000000 -0.250000000 0.125000000
3 0.500000000 -0.500000000 0.500000000 0.062500000
4 0.000000000 0.500000000 0.000000000 0.093750000
5 0.750000000 -0.250000000 0.750000000 0.375000000
6 0.500000000 0.000000000 0.500000000 0.187500000
7 0.000000000 -1.000000000 0.000000000 0.046875000
8 -0.500000000 -1.000000000 0.000000000 0.093750000
(b) 另外一种得到$\mathbf{q}$点权重的方式是用PW/tools/kpoints.x,kpoints.x的运行是交互式的,如下所示:
***************************************************
* *
* Welcome to the special points world! *
*________________________________________________ *
* 1 = cubic p (sc ) 8 = orthor p (so ) *
* 2 = cubic f (fcc) 9 = orthor base-cent. *
* 3 = cubic i (bcc) 10 = orthor face-cent. *
* 4 = hex & trig p 11 = orthor body-cent. *
* 5 = trigonal r 12 = monoclinic p *
* 6 = tetrag p (st ) 13 = monocl base-cent. *
* 7 = tetrag i (bct) 14 = triclinic p *
***************************************************
bravais lattice >> 2
filout [mesh_k] >> mesh_k
mesh: n1 n2 n3 >> 4 4 4
mesh: k1 k2 k3 (0 no shift, 1 shifted) >> 0 0 0
write all k? [f] >> f
# of k-points == 8 of 64
此时目录中会生成info和mesh_k两个文件。其中mesh_k内容如下:
8
1 0.0000000 0.0000000 0.0000000 1.00
2 -0.2500000 -0.2500000 0.2500000 8.00
3 -0.5000000 -0.5000000 0.5000000 4.00
4 0.0000000 0.0000000 0.5000000 6.00
5 -0.2500000 -0.2500000 0.7500000 24.00
6 -0.5000000 -0.5000000 1.0000000 12.00
7 0.0000000 0.0000000 1.0000000 3.00
8 -0.5000000 0.0000000 1.0000000 6.00
用在下面的ph.x输入中
Electron-phonon coefficients
&inputph
tr2_ph=1.0d-10,
prefix='pwscf',
fildvscf='pbdv',
amass(1)=207.2,
outdir='./tmp',
fildyn='pb.dyn',
electron_phonon='interpolated',
el_ph_sigma=0.005,
el_ph_nsigma=10,
trans=.true.,
ldisp=.false.
q_in_band_form=.false.
/
0.0000000 0.0000000 0.0000000
其中$\mathbf{q}$点一次只能输入一个点,所以需要单独计算8次ph.x,其余7个$\mathbf{q}$点如下:
-0.2500000 -0.2500000 0.2500000
-0.5000000 -0.5000000 0.5000000
0.0000000 0.0000000 0.5000000
-0.2500000 -0.2500000 0.7500000
-0.5000000 -0.5000000 1.0000000
0.0000000 0.0000000 1.0000000
-0.5000000 0.0000000 1.0000000
(4) q2r.x变换力常数
&input
zasr='simple', fildyn='pb.dyn', flfrc='pb444.fc', la2F=.true.
/
在输出文件q2r.out中需要有以下内容。
fft-check success (sum of imaginary terms < 10^-12)
(5) matdyn.x计算q路径上声子线宽
设置la2F=.true., dos=.false.会计算声子线宽$\gamma$,$\mathbf{q}$点路径是笛卡尔坐标,以2$\pi$/alat为单位。这里的q点路径,请参考上文第4.2节的声子色散路径设置,这里只取了比较稀疏的19个点作为示意。
&input
asr='simple', amass(1)=207.2,
flfrc='pb444.fc', flfrq='pb444.freq', la2F=.true., dos=.false.
/
19
0.000 0.0 0.0 0.0
0.125 0.0 0.0 0.0
0.250 0.0 0.0 0.0
0.375 0.0 0.0 0.0
0.500 0.0 0.0 0.0
0.750 0.0 0.0 0.0
1.000 0.0 0.0 0.0
0.825 0.125 0.125 0.0
0.750 0.250 0.250 0.0
0.625 0.375 0.375 0.0
0.500 0.500 0.500 0.0
0.325 0.325 0.325 0.0
0.250 0.250 0.250 0.0
0.125 0.125 0.125 0.0
0.000 0.000 0.000 0.0
0.125 0.125 0.000 0.0
0.250 0.250 0.000 0.0
0.325 0.325 0.000 0.0
0.500 0.500 0.000 0.0
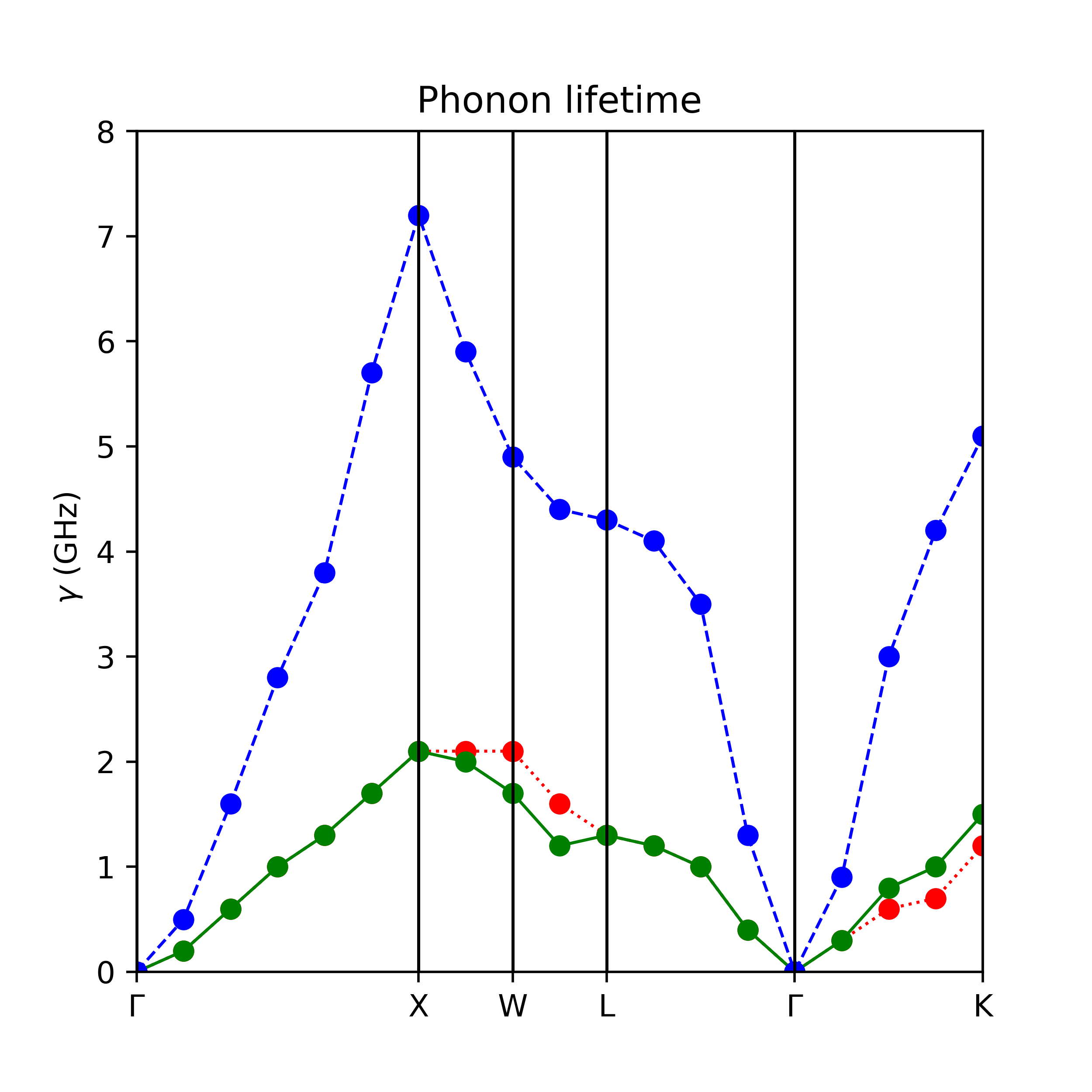
得到gam.lines文件画图如下,文件包含了3条声子色散在各个$\mathbf{q}$点的声子线宽,其中展宽取最后收敛的degauss=0.05。
(6) matdyn.x计算 $\lambda$ 和 $\alpha^{2} F(\omega)$
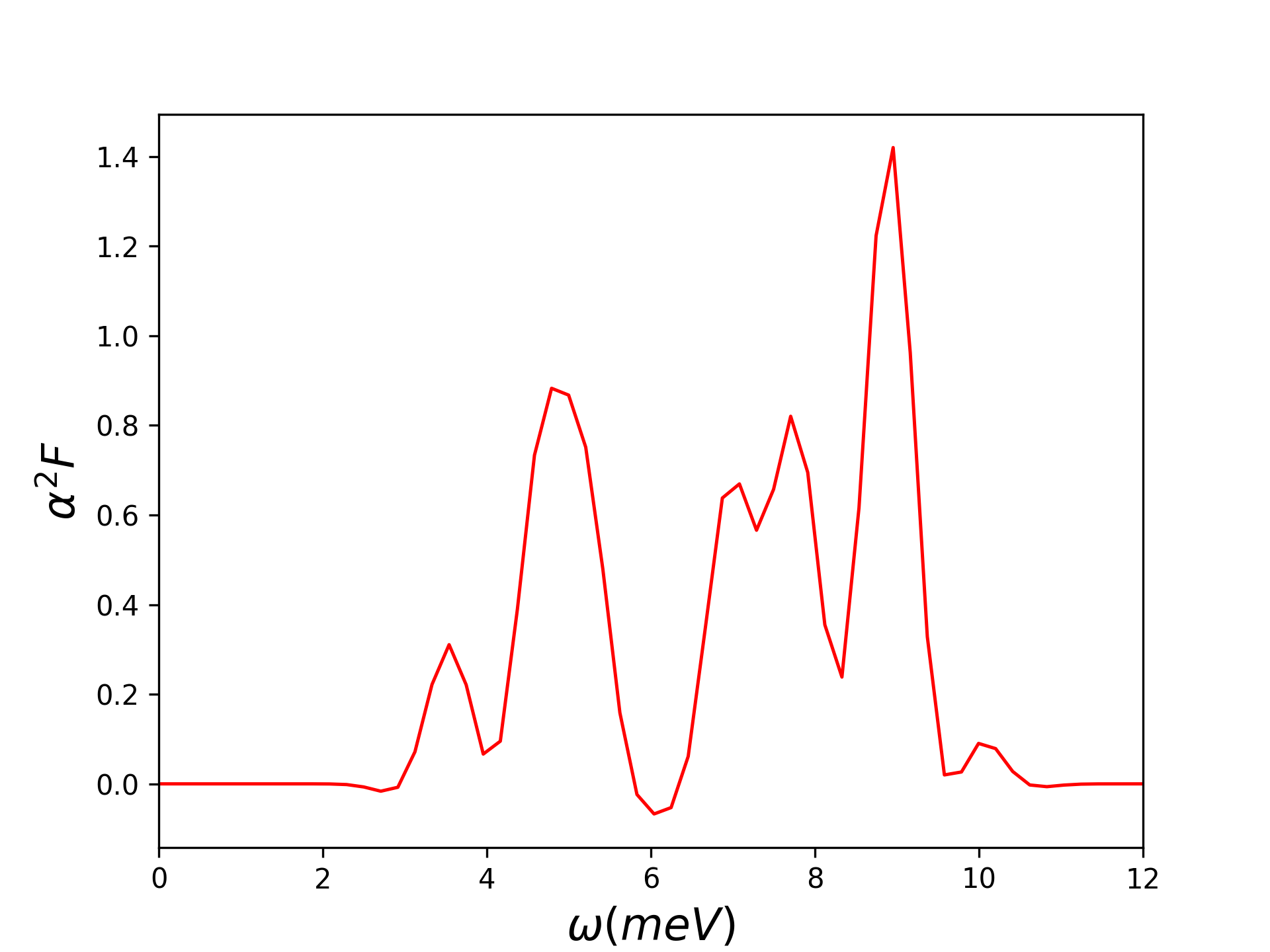
设置la2F=.true., dos=.true.,计算电子声子耦合常数$\lambda$和Eliashberg谱函数$\alpha^{2} F(\omega)$,输出’phonon.dos’声子态密度文件。
&input
asr='simple', amass(1)=207.2,
flfrc='pb444.fc', flfrq='pb444.freq', la2F=.true., dos=.true.
fldos='phonon.dos', nk1=10, nk2=10, nk3=10, ndos=50
/
(7) lambda.x计算 $\lambda$ 和 $T_C$
计算电子声子耦合常数$\lambda$和McMillan公式中的超导转变温度$T_C$。$\mu^{∗}$是一个描述库仑屏蔽作用的经验常数,取值常在0.1到0.16之间,这里取值0.1。这里的8个$\mathbf{q}$点坐标及权重取自第(3)步的ph.x输出即(a)方法。
10 0.12 1 ! emax (something more than highest phonon mode in THz), degauss, smearing method
8 ! Number of q-points for which EPC is calculated,
0.000000000 0.000000000 0.000000000 0.015625000
-0.250000000 0.250000000 -0.250000000 0.125000000
0.500000000 -0.500000000 0.500000000 0.062500000
0.000000000 0.500000000 0.000000000 0.093750000
0.750000000 -0.250000000 0.750000000 0.375000000
0.500000000 0.000000000 0.500000000 0.187500000
0.000000000 -1.000000000 0.000000000 0.046875000
-0.500000000 -1.000000000 0.000000000 0.093750000
elph_dir/elph.inp_lambda.1 ! elph output file names,
elph_dir/elph.inp_lambda.2 ! in the same order as the q-points before
elph_dir/elph.inp_lambda.3
elph_dir/elph.inp_lambda.4
elph_dir/elph.inp_lambda.5
elph_dir/elph.inp_lambda.6
elph_dir/elph.inp_lambda.7
elph_dir/elph.inp_lambda.8
0.10 ! \mu the Coloumb coefficient in the modified
! Allen-Dynes formula for T_c (via \omega_log)
或使用(3)中的(b)方法,也就是kpoints.x生成$\mathbf{q}$点,
10 0.12 1 ! emax (something more than highest phonon mode in THz), degauss, smearing method
8 ! Number of q-points for which EPC is calculated,
0.0000000 0.0000000 0.0000000 1.00 ! the first q-point, use kpoints.x program to calculate
-0.2500000 -0.2500000 0.2500000 8.00 ! q-points and their weight
-0.5000000 -0.5000000 0.5000000 4.00 !
0.0000000 0.0000000 0.5000000 6.00 ! 4th q-point, qx,qy,qz
-0.2500000 -0.2500000 0.7500000 24.00 !
-0.5000000 -0.5000000 1.0000000 12.00 !
0.0000000 0.0000000 1.0000000 3.00 !
-0.5000000 0.0000000 1.0000000 6.00 ! the last q-point
elph_dir/elph.inp_lambda.1 ! elph output file names,
elph_dir/elph.inp_lambda.2 ! in the same order as the q-points before
elph_dir/elph.inp_lambda.3
elph_dir/elph.inp_lambda.4
elph_dir/elph.inp_lambda.5
elph_dir/elph.inp_lambda.6
elph_dir/elph.inp_lambda.7
elph_dir/elph.inp_lambda.8
0.10 ! \mu the Coloumb coefficient in the modified
! Allen-Dynes formula for T_c (via \omega_log)
lambda.x的输出内容如下:
lambda = 1.005300 ( 1.005226 ) <log w>= 76.194 K N(Ef)= 3.393003 at degauss= 0.005
lambda = 0.922870 ( 0.922796 ) <log w>= 74.369 K N(Ef)= 3.306111 at degauss= 0.010
lambda = 0.925753 ( 0.925674 ) <log w>= 73.184 K N(Ef)= 3.309101 at degauss= 0.015
lambda = 0.913178 ( 0.913101 ) <log w>= 73.048 K N(Ef)= 3.317735 at degauss= 0.020
lambda = 0.903445 ( 0.903370 ) <log w>= 73.032 K N(Ef)= 3.330436 at degauss= 0.025
lambda = 0.896805 ( 0.896730 ) <log w>= 72.978 K N(Ef)= 3.343493 at degauss= 0.030
lambda = 0.892266 ( 0.892192 ) <log w>= 72.911 K N(Ef)= 3.356210 at degauss= 0.035
lambda = 0.889445 ( 0.889373 ) <log w>= 72.844 K N(Ef)= 3.368160 at degauss= 0.040
lambda = 0.888256 ( 0.888184 ) <log w>= 72.774 K N(Ef)= 3.379120 at degauss= 0.045
lambda = 0.888413 ( 0.888341 ) <log w>= 72.695 K N(Ef)= 3.389104 at degauss= 0.050
lambda omega_log T_c
1.00530 76.194 5.349
0.92287 74.369 4.549
0.92575 73.184 4.500
0.91318 73.048 4.388
0.90345 73.032 4.306
0.89680 72.978 4.248
0.89227 72.911 4.206
0.88945 72.844 4.178
0.88826 72.774 4.165
0.88841 72.695 4.161
这里各个计算量的值在degauss=0.015接近收敛,得到$T_c$=4.500K。
得到输出文件alpha2F.dat,取最后一列展宽为0.05的结果画图如下。
本节输入输出文件见这里。
4.3.2 使用EPW模块计算电子-声子耦合系数
电子-声子耦合系数对于布里渊区取网格的密度收敛十分缓慢,即需要很密的点,上一节的结果实际上对于$\mathbf{k}$点还没有完全收敛。EPW使用拟合到的Wannier局域轨道来计算电子-声子耦合系数,这也是原子较多体系可行的方案。
5. 杂化泛函和Wannier函数
HSE06是一种杂化泛函(Hybrid functional),其中交换作用短程部分用GGA和Hatree-Fock的混合,长程部分使用GGA,关联则统一使用GGA。HSE06相对于GGA计算结果,总能、力和能带的准确度有系统性的提升[12],但是计算量很大。Wannier基函数是与平面波基函数可以相互变换的完备基,是一种局域轨道基函数[13]。QE中的wannier模块可以提取$\mathbf{k}$点网格上的平面波波函数结果,得到最局域万尼尔函数MLWF,并使用MLWF快速计算出更多$\mathbf{k}$点的能级。
计算前需要编译wannier90模块,QE目录运行make w90。
计算HSE能带有以下四步:
- 用pw.x运行‘scf’。
- 运行wannier90.x -pp (预处理pre-process,或在输入文件内写postproc_setup = .true.)生成seedname.nnkp。
- 运行pw2wannier90.x读入pw输出文件、seedname.pw2wan和seedname.nnkp, 生成seedname.mmn, seedname.amn和seedname.eig。
- 运行wannier90.x得到能带。
5.1 杂化泛函自洽计算
用HSE06进行scf计算,运行pw.x,输入如下,这里采用了pslibrary1.0的超软赝势,采用了PBE的晶格常数(见第1.4节),也可以用HSE06做vc-relax或状态方程计算,计算需要关掉对称性,得到完整的k网格(4×4×4=64个$\mathbf{k}$点):
&CONTROL
calculation='scf', disk_io='low', prefix='pwscf',
pseudo_dir='./', outdir='./tmp', verbosity='high'
tprnfor=.true., tstress=.true., forc_conv_thr=1.0d-5
/
&SYSTEM
ibrav= 0,
nat= 2, ntyp= 2,
occupations = 'smearing', smearing = 'gauss', degauss = 1.0d-9
ecutwfc= 50, ecutrho = 500,
input_dft='hse'
exxdiv_treatment = 'gygi-baldereschi'
ecutvcut = 0.7
x_gamma_extrapolation = .true.
nqx1=4, nqx2=4, nqx3=4,
nosym=.true.
nosym_evc=.true.
noinv=.true.
/
&ELECTRONS
electron_maxstep = 100
conv_thr = 1.0d-9
mixing_mode = 'plain'
mixing_beta = 0.8d0
diagonalization = 'david'
/
&IONS
ion_dynamics='bfgs'
/
&CELL
press_conv_thr=0.1
/
ATOMIC_SPECIES
Si 28.08550 Si.UPF
C 12.01070 C.UPF
ATOMIC_POSITIONS (crystal)
Si 0.000000000 0.000000000 0.000000000
C 0.250000000 0.250000000 0.250000000
K_POINTS {automatic}
4 4 4 0 0 0
CELL_PARAMETERS (angstrom)
2.188890473 2.188890473 0.000000000
0.000000000 2.188890473 2.188890473
2.188890473 0.000000000 2.188890473
在新版本(>=6.4)中加入了localization_thr参数,设置为0.004,限于模守恒赝势,对于大体系(超胞)可以减少计算量[19],非$\Gamma$点功能还处于试验阶段。
5.2 提取Wannier函数并计算能带
用wannier.x模块作为上一节HSE自洽计算的后处理,生成能带图。本节介绍的方法也适用于其他DFT计算,如PBE的能带。
输入文件sc.win如下,其中mp_grid = 4 4 4要与HSE06的scf计算中$\mathbf{k}$点一致,kpoints采用上一节scf计算pw.x输出的$\mathbf{k}$点拷贝过来(分数坐标,见wannier90 user_guide 2.4.6节),运行wannier90.x -pp sc:
num_bands = 8
num_wann = 8
dis_win_max = 17.0d0
dis_froz_max = 6.4d0
dis_num_iter = 120
dis_mix_ratio = 1.d0
num_iter = 50
num_print_cycles = 10
Begin Atoms_Frac
Si 0.000000000 0.000000000 0.000000000
C 0.250000000 0.250000000 0.250000000
End Atoms_Frac
Begin Projections
Si : sp3
C : sp3
End Projections
begin kpoint_path
L 0.50000 0.50000 0.5000 G 0.00000 0.00000 0.0000
G 0.00000 0.00000 0.0000 X 0.50000 0.00000 0.5000
X 0.50000 -0.50000 0.0000 K 0.37500 -0.37500 0.0000
K 0.37500 -0.37500 0.0000 G 0.00000 0.00000 0.0000
end kpoint_path
!bands_plot = .true.
Begin Unit_Cell_Cart
2.188890473 2.188890473 0.000000000
0.000000000 2.188890473 2.188890473
2.188890473 0.000000000 2.188890473
End Unit_Cell_Cart
mp_grid = 4 4 4
begin kpoints
0.0000000 0.0000000 0.0000000
0.0000000 0.0000000 0.2500000
0.0000000 0.0000000 -0.5000000
0.0000000 0.0000000 -0.2500000
0.0000000 0.2500000 0.0000000
0.0000000 0.2500000 0.2500000
0.0000000 0.2500000 -0.5000000
0.0000000 0.2500000 -0.2500000
0.0000000 -0.5000000 0.0000000
0.0000000 -0.5000000 0.2500000
0.0000000 -0.5000000 -0.5000000
0.0000000 -0.5000000 -0.2500000
0.0000000 -0.2500000 0.0000000
0.0000000 -0.2500000 0.2500000
0.0000000 -0.2500000 -0.5000000
0.0000000 -0.2500000 -0.2500000
0.2500000 0.0000000 0.0000000
0.2500000 0.0000000 0.2500000
0.2500000 0.0000000 -0.5000000
0.2500000 0.0000000 -0.2500000
0.2500000 0.2500000 0.0000000
0.2500000 0.2500000 0.2500000
0.2500000 0.2500000 -0.5000000
0.2500000 0.2500000 -0.2500000
0.2500000 -0.5000000 0.0000000
0.2500000 -0.5000000 0.2500000
0.2500000 -0.5000000 -0.5000000
0.2500000 -0.5000000 -0.2500000
0.2500000 -0.2500000 0.0000000
0.2500000 -0.2500000 0.2500000
0.2500000 -0.2500000 -0.5000000
0.2500000 -0.2500000 -0.2500000
-0.5000000 0.0000000 0.0000000
-0.5000000 0.0000000 0.2500000
-0.5000000 0.0000000 -0.5000000
-0.5000000 0.0000000 -0.2500000
-0.5000000 0.2500000 0.0000000
-0.5000000 0.2500000 0.2500000
-0.5000000 0.2500000 -0.5000000
-0.5000000 0.2500000 -0.2500000
-0.5000000 -0.5000000 0.0000000
-0.5000000 -0.5000000 0.2500000
-0.5000000 -0.5000000 -0.5000000
-0.5000000 -0.5000000 -0.2500000
-0.5000000 -0.2500000 0.0000000
-0.5000000 -0.2500000 0.2500000
-0.5000000 -0.2500000 -0.5000000
-0.5000000 -0.2500000 -0.2500000
-0.2500000 0.0000000 0.0000000
-0.2500000 0.0000000 0.2500000
-0.2500000 0.0000000 -0.5000000
-0.2500000 0.0000000 -0.2500000
-0.2500000 0.2500000 0.0000000
-0.2500000 0.2500000 0.2500000
-0.2500000 0.2500000 -0.5000000
-0.2500000 0.2500000 -0.2500000
-0.2500000 -0.5000000 0.0000000
-0.2500000 -0.5000000 0.2500000
-0.2500000 -0.5000000 -0.5000000
-0.2500000 -0.5000000 -0.2500000
-0.2500000 -0.2500000 0.0000000
-0.2500000 -0.2500000 0.2500000
-0.2500000 -0.2500000 -0.5000000
-0.2500000 -0.2500000 -0.2500000
End Kpoints
以下文件保存为pw2wan.inp,运行pw2wannier90.x < pw2wan.inp > pw2wan.out:
&inputpp
outdir = './tmp'
prefix = 'pwscf'
seedname = 'sc'
spin_component = 'none'
write_mmn = .true.
write_amn = .true.
write_unk = .true.
/
将sc.win中的!bands_plot = .true.注释叹号删去,再运行一次wannier90.x sc,得到wannier基函数的能带值sc_band.dat,用这个文件画图,gnuplot输入文件如下:
reset
set terminal pngcairo size 580,880 enhanced font 'Times-Roman,15'
set output "out.png"
set style data dots
set nokey
set label "SiC HSE06 Eg=2.15eV" at graph 0.05,graph 0.96
set xrange [0: 4.70794]
!set yrange [ -20.0 : 20.0]
set yrange [ -20 : 20]
set arrow from 1.24296, -20.0 to 1.24296, 20.0 nohead
set arrow from 2.67820, -20.0 to 2.67820, 20.0 nohead
set arrow from 3.18564, -20.0 to 3.18564, 20.0 nohead
set xtics ("L" 0.00000,"{/Symbol G}" 1.24296,"X" 2.67820,"K" 3.18564,"{/Symbol G}" 4.70794)
plot "sc_band.dat" using 1:($2-0.88318010E+01) with lines lt 1 lw 2 lc rgb "red"
这里参考安装包中的例子q-e-qe-6.3/PW/examples/EXX_example以及q-e-qe-6.3/W90/examples/example03和example05。用8×8×8 $\mathbf{k}$点的输入及输出文件见这里,供参考。
注:(1)这里作为示例取$\mathbf{k}$点偏小了,实际计算建议$\mathbf{k}$点增加到6×6×6以上,wannier拟合效果好一些;(2)HSE自洽scf似乎可以不关对称性用较少的$\mathbf{k}$点,接着做一次scf的完整k网格计算,读取电荷,可以减小部分计算量(QE不支持HSE-nscf);(3)wannier只能选取部分价带导带拟合,含有半芯态的赝势,需要用exclude_bands排除较低的半芯态和较高的能带;(4)Projections一般根据价态电子选择,见wannier手册第三章,不限于原子成键电子的spd类型,有一定的任意性,对于同一种材料可能会有不同的选择,但是有些选择得到的能带和scf计算得到的更接近;(5)可以将wannier中心设置在其他位置,如键中心等;(6)num_wann等于Projections设置的总的轨道数;(7)num_bands等于或大于num_wann,num_bands加上exclude_bands个数等于scf计算中的nbnd,一个wannier轨道形成一条带,num_bands大于num_wann时,多出来的带默认会disentangle,设置dis_win_min和dis_win_max作为能量窗口。(8)如果投影出问题,尝试使用auto_projections = .true.,并注释Projections。
结果如下图,带隙值与PBE(见1.4节)相比有所提升。
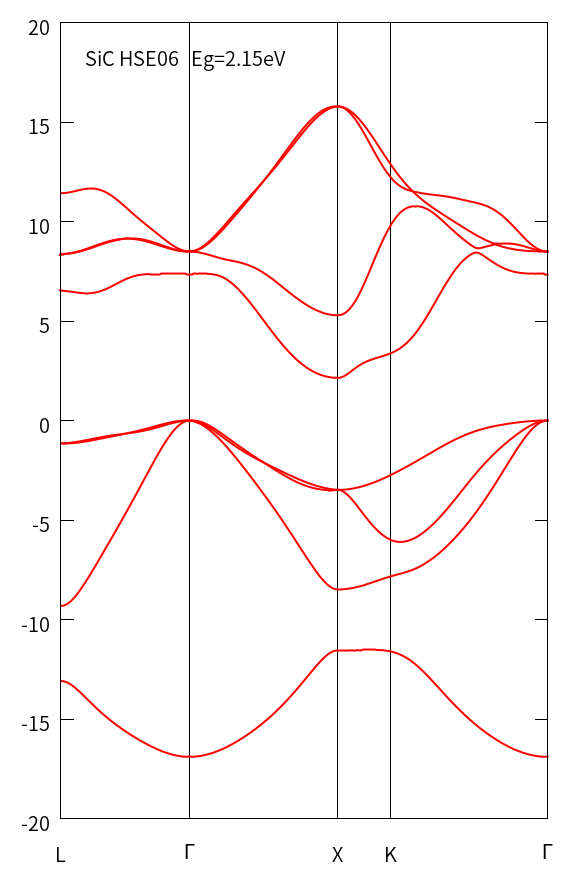
5.3 HSE自洽计算能带
以上HSE自洽计算——平面波转换为wannier波函数——生成能带的步骤,是QE推荐的方法。此外,参考exabyte的做法,在scf的$\mathbf{k}$点网格基础上追加一些权重几乎为0的特殊路径$\mathbf{k}$点(不能等于0,权重大于$1\times10^{-7}$),这样仅做一次scf计算就得到能带,这种方法比较接近VASP计算HSE能带时的方式。
经过测试,这种方法需要设置nqx1, nqx2, nqx3=1,造成EXX的布里渊区积分只用了$\Gamma$点,这对于超胞的误差较小。但是,原胞计算误差就难以忽略,不推荐使用了。经过测试,这样计算SiC的能带,带隙在3.4-3.8eV,而5.1节中nqx1, nqx2, nqx3=4,带隙为2.15eV,QE程序中的机制造成HSE计算要求nqx的网格与任意$\mathbf{k}$点的和包含在和$\mathbf{k}$点网格中,这么强的限制造成除了nqx1, nqx2, nqx3=1以外的设置没法实现,通过增加$\mathbf{k}$点网格的密度也不能消除,所以,目前还没办法解决这个问题,目前用scf计算追加$\mathbf{k}$点的方法只适用尺寸较大的超胞,这时,只用$\Gamma$点近似布里渊区的积分。
6. 波函数投影
QE计算的波函数是以平面波基组表示的,而原子轨道有更好的直观性和可解释性,是分析能带不可或缺的手段,可以将各个$\mathbf{k}$点的各个能级的波函数,投影到以各个原子坐标为中心的s、p、d、f等球谐函数上(以及非共线和磁性计算中的自旋),即为(local)projected density of states,用projwfc.x模块计算,下面以$SnO_{2}$(rutile)的投影态密度为例。投影计算前可以检查一下赝势文件中PSWFC部分内容非空,否则无法使用projwfc.x模块。
6.1 投影态密度
做vc-relax,输入文件如下:
&CONTROL
calculation='vc-relax', disk_io='low', prefix='pwscf',
pseudo_dir='./', outdir='./tmp', verbosity='high'
tprnfor=.true., tstress=.true., forc_conv_thr=1.0d-5, etot_conv_thr=1.0d-5, nstep=100,
/
&SYSTEM
ibrav= 6,
celldm(1)=8.952142,
celldm(3)=0.672620,
nat= 6,
ntyp= 2,
occupations = 'fixed', nbnd=32,
ecutwfc= 50, ecutrho = 600,
/
&ELECTRONS
electron_maxstep = 100, conv_thr = 1.0d-9, mixing_mode = 'plain'
mixing_beta = 0.7d0, diagonalization = 'david'
/
&IONS
ion_dynamics='bfgs'
/
&CELL
press = 0.00 ,
press_conv_thr=0.1
cell_dynamics = 'bfgs' ,
/
ATOMIC_SPECIES
Sn 118.710 Sn.pbe-dn-rrkjus_psl.1.0.0.UPF
O 15.999 O.pbe-n-rrkjus_psl.1.0.0.UPF
ATOMIC_POSITIONS (crystal)
Sn 0.000000 0.000000 0.000000
Sn 0.500000 0.500000 0.500000
O 0.307000 0.307000 0.000000
O -0.307000 -0.307000 0.000000
O 0.193000 0.807000 0.500000
O 0.807000 0.193000 0.500000
K_POINTS {automatic}
6 6 9 0 0 0
在输出中可以找到
point group D_4h(4/mmm)
说明对称性是正确的,注意到这个材料在QE6.3之前找到的结果是D_2h。vc-relax结果是
Begin final coordinates
new unit-cell volume = 513.62842 a.u.^3 ( 76.11188 Ang^3 )
density = 6.57602 g/cm^3
CELL_PARAMETERS (alat= 8.95214200)
1.021642232 0.000000000 0.000000000
0.000000000 1.021642232 0.000000000
0.000000000 0.000000000 0.685915293
ATOMIC_POSITIONS (crystal)
Sn 0.000000000 0.000000000 -0.000000000
Sn 0.500000000 0.500000000 0.500000000
O 0.306642111 0.306642111 -0.000000000
O -0.306642111 -0.306642111 -0.000000000
O 0.193357889 0.806642111 0.500000000
O 0.806642111 0.193357889 0.500000000
End final coordinates
计算DOS需要较密的$\mathbf{k}$点,再做一次nscf计算,$\mathbf{k}$点设置为
K_POINTS {automatic}
13 13 20 0 0 0
以下文件保存为proj.inp,运行mpirun projwfc.x <proj.inp >proj.out。
&PROJWFC
prefix='pwscf',
outdir='./tmp',
ngauss=0,
degauss=0.01,
DeltaE=0.005
lsym=.true.
filpdos='sno',
filproj='sno',
/
用以下Python程序画图:
#!/bin/env python
# -*- coding: utf-8 -*-
"""
@author: yyyu200@163.com
"""
import matplotlib as mpl
mpl.use('Agg')
import matplotlib.pyplot as plt
import numpy as np
lw=1.2
F=plt.gcf()
F.clf()
p=plt.subplot(1, 1, 1)
# change by-hand here, read scf input in the future
elem=['Sn','O']
ielem=np.array([2,4],dtype=np.int32) # number of atoms for each element
orb=[['s','p','d'],['s','p']] # projectors for each element
iorb=np.array([3,2],dtype=np.int32) # number of projectors for each element
num_file=np.dot(ielem,iorb)
nat=np.sum(ielem)
D=[]
N=len(elem)
#scf ATOMIC_POSITIONS should be sorted in the same order as above
count=0
count_at=0
for n in range(N):
for i in range(ielem[n]):
for j in range(iorb[n]):
print(n,i,j,count_at+1,elem[n],j+1,orb[n][j])
fname='sno.pdos_atm#{}({})_wfc#{}({})'.format(count_at+1,elem[n],j+1,orb[n][j])
D.append(np.loadtxt(fname,dtype=np.float32))
count+=1
count_at+=1
e_fermi=8.7233
line1=plt.plot(D[0][:,0]-e_fermi,D[0][:,1]+D[3][:,1],color='g',linewidth=lw,label='Sn s' )
line2=plt.plot(D[0][:,0]-e_fermi,D[1][:,1]+D[4][:,1],color='r',linewidth=lw,label='Sn p' )
line3=plt.plot(D[0][:,0]-e_fermi,D[2][:,1]+D[5][:,1],color='b',linewidth=lw,label='Sn d' )
line4=plt.plot(D[0][:,0]-e_fermi,D[6][:,1]+D[8][:,1]+D[10][:,1]+D[12][:,1],color='cyan',linewidth=lw,label='O s' )
line5=plt.plot(D[0][:,0]-e_fermi,D[7][:,1]+D[9][:,1]+D[11][:,1]+D[13][:,1],color='grey',linewidth=lw,label='O p' )
plt.xlim([-10,7])
plt.ylim([0,10.5])
plt.ylabel(r'DOS (a.u.)',fontsize=16)
plt.xlabel(r'E (eV) ',fontsize=16)
plt.legend()
plt.savefig('pdos.png',dpi=200)
结果如图所示。
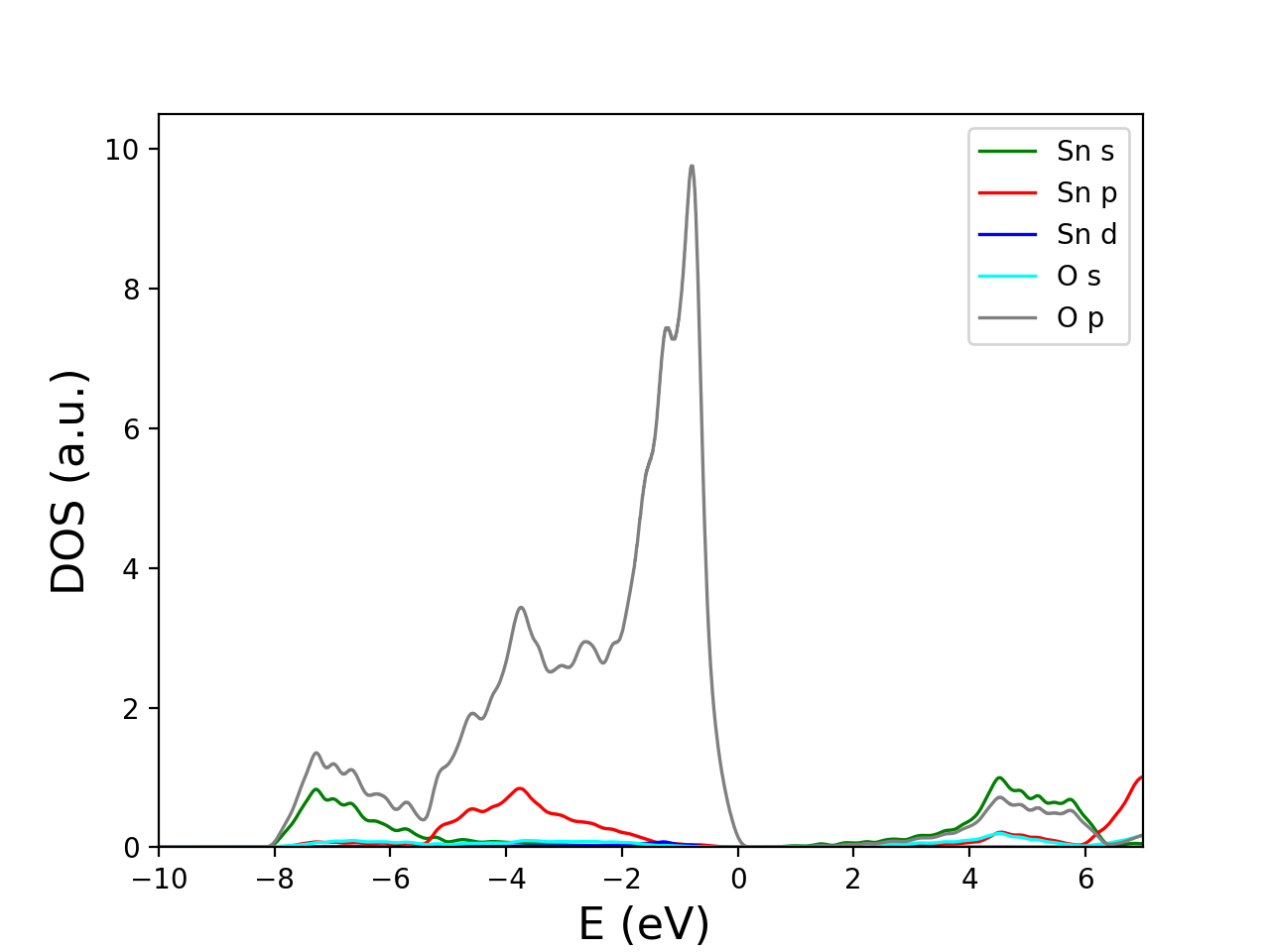
6.2 投影能带
(1)运行pw.x,进行一次能带计算,输入中修改calculation='bands',$\mathbf{k}$点设置为:
K_POINTS {crystal_b}
3
M 30
gG 30
Z 0
(2)运行projwfc.x,输入同上,此时读入波函数(注意应该是能带计算波函数),计算投影。
(3)用projwfc.x生成的sno.projwfc_up文件,可以和bands.x生成的bd.dat文件,一起作为输入,画出各个能带的原子轨道投影(步骤见1.4节),画图脚本如下(更新的代码见这里):
#!/usr/bin/env python3
# -*- coding: utf-8 -*-
import numpy as np
def parse_filband(feig, npl=10):
# feig : filband in bands.x input file
# npl : number per line, 10 for bands.x, 6 for phonon
import re
f=open(feig,'r')
lines = f.readlines()
header = lines[0].strip()
line = header.strip('\n')
shape = re.split('[,=/]', line)
nbnd = int(shape[1])
nks = int(shape[3])
eig = np.zeros((nks, nbnd+1), dtype=np.float32)
dividend = nbnd
divisor = npl
div = nbnd // npl + 1 if nbnd % npl == 0 else nbnd // npl + 2
kinfo=[]
for index, value in enumerate(lines[1:]):
value = value.strip(' \n')
quotient = index // div
remainder = index % div
if remainder == 0:
kinfo.append(value)
else:
value = re.split('[ ]+', value)
a = (remainder - 1) * npl
b = a + len(value)
eig[quotient][a:b] = value
f.close()
return eig, nbnd, nks, kinfo
def match_pattern_with_line_numbers(pattern, text):
import re
line_number = 1
for line in text.splitlines():
match = re.search(pattern, line)
if match:
yield line_number, match
line_number += 1
def get_sum_orbit(orb, ntype, ielem, i_soc):
iorb=np.zeros([ntype,],dtype=np.int32) # number of projectors for each element
for i in range(ntype):
iorb[i]=len(orb[i])
lorb=np.zeros([ntype,],dtype=np.int32) # number of local orbital for each element
for i in range(ntype):
for j in orb[i]:
if j == 's':
lorb[i]+=1*i_soc
elif j == 'p':
lorb[i]+=3*i_soc
elif j == 'd':
lorb[i]+=5*i_soc
elif j == 'f':
lorb[i]+=7*i_soc
else:
print("unexpect: ",j)
assert False
return np.dot(ielem,lorb)
def draw_proj_band(proj_file, fig_file, eig, nbnd, nks, kinfo, dpi = 500):
# oo, orbital index for each kind of color, oo can be generated by the following commands
#grep '[a-zA-Z]' sno.projwfc_up |grep 2P|awk '{printf( $1-1",")}'
oo=[[0,9], # s of Sn atoms 1,2
[1,2,3,10,11,12], # pz,px,py of Sn atoms 1,2
[4,5,6,7,8,13,14,15,16,17], # dz2, dzx, dzy, dx2-y2, dxy of Sn atoms 1,2
[18,22,26,30], # s of O atoms 1,2,3,4
[19,20,21,23,24,25,27,28,29,31,32,33]] # pz,px,py of O atoms 1,2,3,4
nband=26 # highest valence band for ref
eig_ref=max(eig[:, nband-1]) # VBM for insulators
#eig_ref= 0.00 # fermi energy level in scf output for metals
ymin=-23 # plot range for y-axis
ymax=10
lw=0.5 # line width
ielem=np.array([2,4],dtype=np.int32) # number of atoms for each element, 2 Sn atoms and 4 O atoms in the cell
color=['r','g','orange','cyan','blue']
label=['Sn s','Sn p','Sn d','O s','O p']
scale=90.0
x_ticks=[0,30,60]
x_labels=['M', r'${\Gamma}$', 'Z']
# end of (the most often) parameters
ntype=len(ielem) # number of atom types
nat = sum(ielem)
nline_io_header=0 # line number at ' F F' or ' T T'
with open(proj_file,'r') as f:
for line_number in range(nat+ntype+6+3):
line = f.readline()
pattern = r" [TF] [TF]"
for i, match in match_pattern_with_line_numbers(pattern, line):
if match.group()[9]=='F':
i_soc = 1
elif match.group()[9]=='T':
i_soc = 2
nline_io_header = line_number+1
assert nline_io_header
#i_soc = 1 # 1: without soc, 2: soc
import matplotlib as mpl
mpl.use('Agg')
import matplotlib.pyplot as plt
F=plt.gcf()
F.set_size_inches([4,5])
p1=plt.subplot(1, 1, 1)
# draw bands
for i in range(nbnd):
line1=plt.plot(np.arange(0,nks), eig[:,i]-eig_ref,color='grey',linewidth=lw )
# draw vertical lines, positions specified by list like [0, 30, 60, ...]
#x_ticks= np.arange(0,nks,30)
for vline in x_ticks:
plt.axvline(x=vline, ymin=ymin, ymax=ymax,linewidth=lw,color='black')
with open(proj_file,'r') as filproj:
for i in range(nline_io_header-1):
l = filproj.readline()
nlorb = int(l.split()[0])
check_nlorb=False
if check_nlorb:
orb=[['s','p','d'],['s','p']] # projectors for each element
_nlorb = get_sum_orbit(orb, ntype, ielem, i_soc)
assert _nlorb==nlorb
print ("summary : ", i_soc, nline_io_header, nlorb, nks, nbnd)
pjmat=np.zeros([nlorb, nks, nbnd], dtype=np.float32)
_ = filproj.readline() # skip ' F F' line
for i in range(nlorb):
_ = filproj.readline() # skip orbit header
for j in range(nks):
for k in range(nbnd):
pjmat[i,j,k]=float(filproj.readline().split()[2])
for i in range(len(oo)): # once plot a type
plt.scatter(-1, ymin-1, 20, c=color[i], alpha=0.5, label=label[i],marker='.',edgecolor='none')# draw an invisible point as the anchor of legend
for k in range(nbnd): # once plot a band
s_of_o=np.zeros([nks,],dtype=np.float32) # size of dots for all kpoints in a band
for j in oo[i]: # gather proj weight for an orbit type, such as all the 'p of Sn', that is pz,px,py of Sn atoms 1,2,...
s_of_o[:]+=pjmat[j,:,k]
plt.scatter(np.arange(0,nks), eig[:,k]-eig_ref, s=scale*s_of_o, c=color[i], alpha=0.5, marker='.',edgecolor='none')
plt.xlim([0,nks-1])
plt.ylim([ymin,ymax])
plt.ylabel(r'E (eV)',fontsize=16)
plt.xticks(x_ticks, x_labels)
plt.subplots_adjust(left=0.20, right=0.75, top=0.95, bottom=0.1)
p1.legend(scatterpoints =1, numpoints=1,markerscale=2.0, bbox_to_anchor=(1.05, 1), loc='upper left', borderaxespad=0.)
plt.savefig(fig_file,dpi=dpi)
if __name__ == '__main__':
_eig, _nbnd, _nks, _kinfo = parse_filband("bd.dat")
draw_proj_band("sno.projwfc_up","pjband.png", _eig, _nbnd, _nks, _kinfo)
print("done")
结果如图所示。
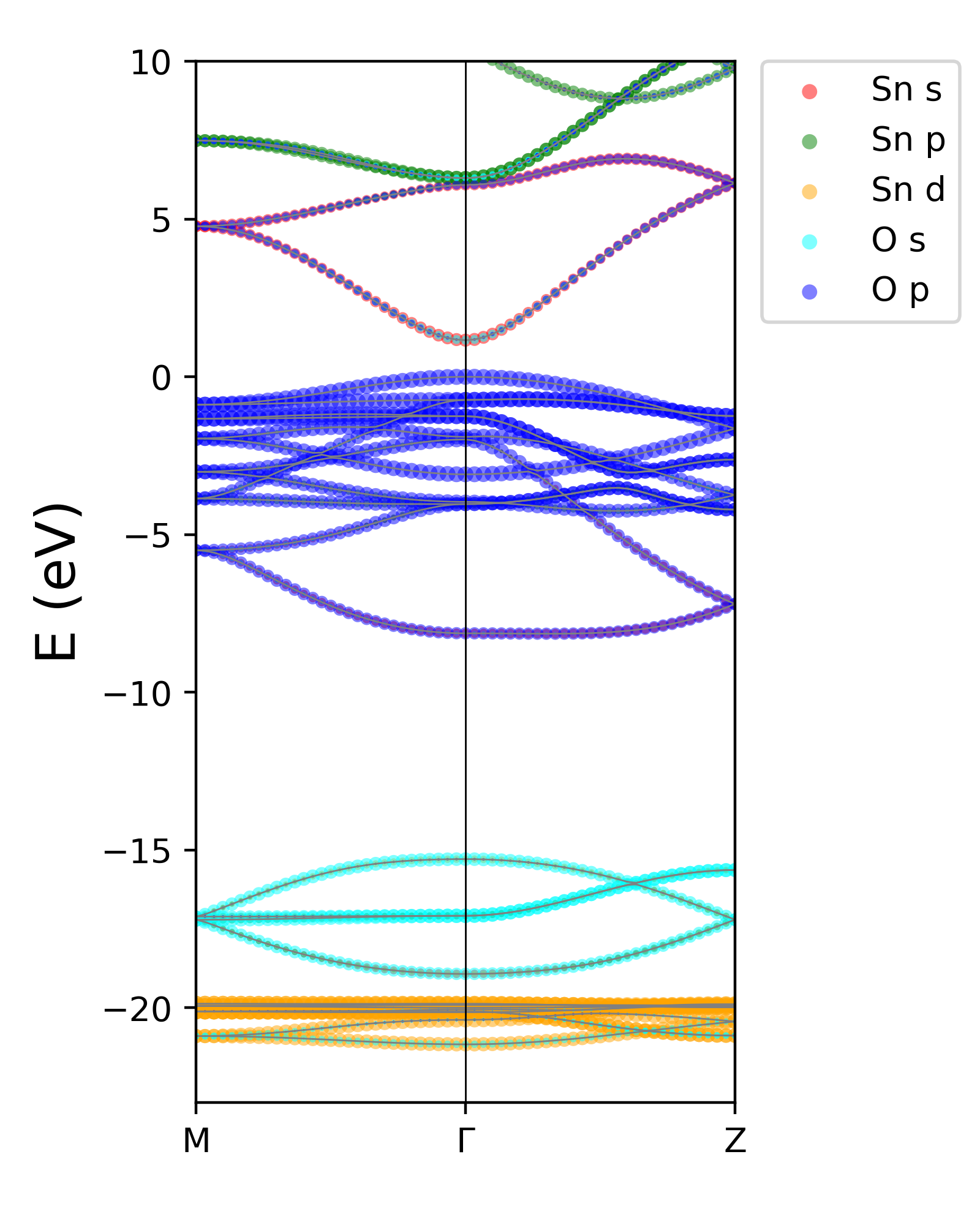
脚本使用说明:
(1) 查看轨道种类
首先在终端运行:grep '[a-zA-Z]' sno.projwfc_up
输出了文件中所有包含字母的行,可以看出,Sn有5S,5P,4D轨道,O有2S,2P轨道,轨道的种类5类(当然还可以细分为P的3个小类Pz,Px,Py,和D轨道的5类,顺序见这里),这里选择5类轨道,以不同颜色作投影能带图。
(2) 确定投影能带图中轨道的序号
再运行grep '[a-zA-Z]' sno.projwfc_up |grep 'Sn 5S'|awk '{printf( $1-1",")}END{print("");}'
得到Sn 5S是哪些序号的(注意空格个数),依次找到Sn的5S,5P,4D轨道,O的2S,2P轨道共5种,所以oo设置成:
oo=[[0,9], # s of Sn atoms 1,2
[1,2,3,10,11,12], # pz,px,py of Sn atoms 1,2
[4,5,6,7,8,13,14,15,16,17], # dz2, dzx, dzy, dx2-y2, dxy of Sn atoms 1,2
[18,22,26,30], # s of O atoms 1,2,3,4
[19,20,21,23,24,25,27,28,29,31,32,33]] # pz,px,py of O atoms 1,2,3,4
这里5组轨道,表示画出5种轨道,是根据作图的需要设置的,可以只画其中一部分轨道。
(3) 能量参考值,能量范围
导体根据scf输出费米能级
eig_vbm= 0.00 # fermi energy level in scf output for metals
绝缘体设置nband,nband是最高的价带的序数,设置价带顶作为能量参考点,可以运行以下命令得到:
grep 'number of electrons' nscf_b.out,对于不加soc计算(默认)需要以上数字除以2;
对于加soc计算填入以上数字即可。
nband=26 # highest valence band for ref
eig_vbm=max(eig[:, nband-1])
ymin,ymax,图中y坐标范围
(4) 每种元素的原子个数
需要写出单元中全部原子个数
ielem=np.array([2,4],dtype=np.int32) # number of atoms for each element
(5) 投影能带的颜色、点的大小
color=['r','g','orange','cyan','blue']
label=['Sn s','Sn p','Sn d','O s','O p']
顺序与oo的类别一致,更多颜色名称见(https://matplotlib.org/3.1.0/gallery/color/named_colors.html)。label是相应轨道名称,在画图图例中显示。
投影能带点的大小
scale=90.0
(6) k点路径坐标、名称
x_ticks=(0,30,60)
x_labels=('M', r'${\Gamma}$', 'Z')
(7) 指定文件名
在程序的最后,’sno.projwfc_up’是projwfc.x中根据filproj设置,加上后缀.projwfc_up的输出文件。 ‘bd.dat’是bands.x输入文件中由filband指定的文件名。 ‘pjband.png’是输出图片文件名。
_eig, _nbnd, _nks, _kinfo = parse_filband("bd.dat")
draw_proj_band("sno.projwfc_up","pjband.png", _eig, _nbnd, _nks, _kinfo)
(8) 检查轨道分组是否正确(可选)
设置check_nlorb=True时,写出每种原子的轨道类型,限s、p、d、f,每个元素一个list,顺序与元素ielem保持一致,需要写出赝势含有的所有轨道类型。
orb=[['s','p'],['s','p','s','d']] # projectors for each element
这样就可以确定投影数据pjsum的维度,即波函数投影的轨道数、k点数、能带数。
代码中assert _nlorb==nlorb检查了轨道,即_nlorb应该和.projwfc_up文件第nline_io_header-1行第三个数字一致。
6.3 波函数
根据Bloch定理,非$\Gamma$点的波函数不具有晶格周期性,直接画图一般是做绝对值平方。有自定义的计算,也可能用到波函数。
波函数绝对值平方可以由pp.x得到,其中设置plot_num=7。可选$\mathbf{k}$点及能带。波函数画图可以用Xcrysden或VESTA,需要转格式。
获取实空间的波函数可以用PP模块中的wfck2r.x,
&inputpp
prefix='pwscf',
outdir='./tmp',
first_k=1
last_k=1
first_band=4
last_band=4
/
其中,可以选择$\mathbf{k}$点及能带,默认是所有的$\mathbf{k}$点和所有的能带。输出wfck2r.mat包含了实空间网格上的复数的波函数。
平面波矢量表示(G空间)的波函数在’outdir’/’prefix’.save/wfc[xxx].dat(或hdf5格式,configure使用--with-hdf5),是二进制文件,将Module/io_base.f90做如下修改,可以写出波函数的文本文件,
102,104c102,104
< FORM='unformatted', STATUS = 'unknown' )
< WRITE(iuni) ik, xk, ispin, gamma_only, scalef
< WRITE(iuni) ngw, igwx, npol, nbnd
---
> FORM='formatted', STATUS = 'unknown' )
> WRITE(iuni,*) ik, xk, ispin, gamma_only, scalef
> WRITE(iuni,*) ngw, igwx, npol, nbnd
132,133c132,133
< WRITE(iuni) b1, b2, b3
< WRITE(iuni) itmp(1:3,1:igwx)
---
> WRITE(iuni,*) b1, b2, b3
> WRITE(iuni,*) itmp(1:3,1:igwx)
186c186
< WRITE(iuni) wtmp(1:npol*igwx)
---
> WRITE(iuni,*) wtmp(1:npol*igwx)
7. 自旋
本节只在计算需要考虑相应特性的材料时加入,默认不加。
7.1 自旋轨道耦合
自旋轨道耦合(SOC)计算基于Dirac方程的二分量近似(Foldy-Wouthuysen transformation)。
在输入中加入noncolin=.true., lspinorb=.true.,。
lspinorb=.true.要求使用至少一种元素的full relativistic赝势。
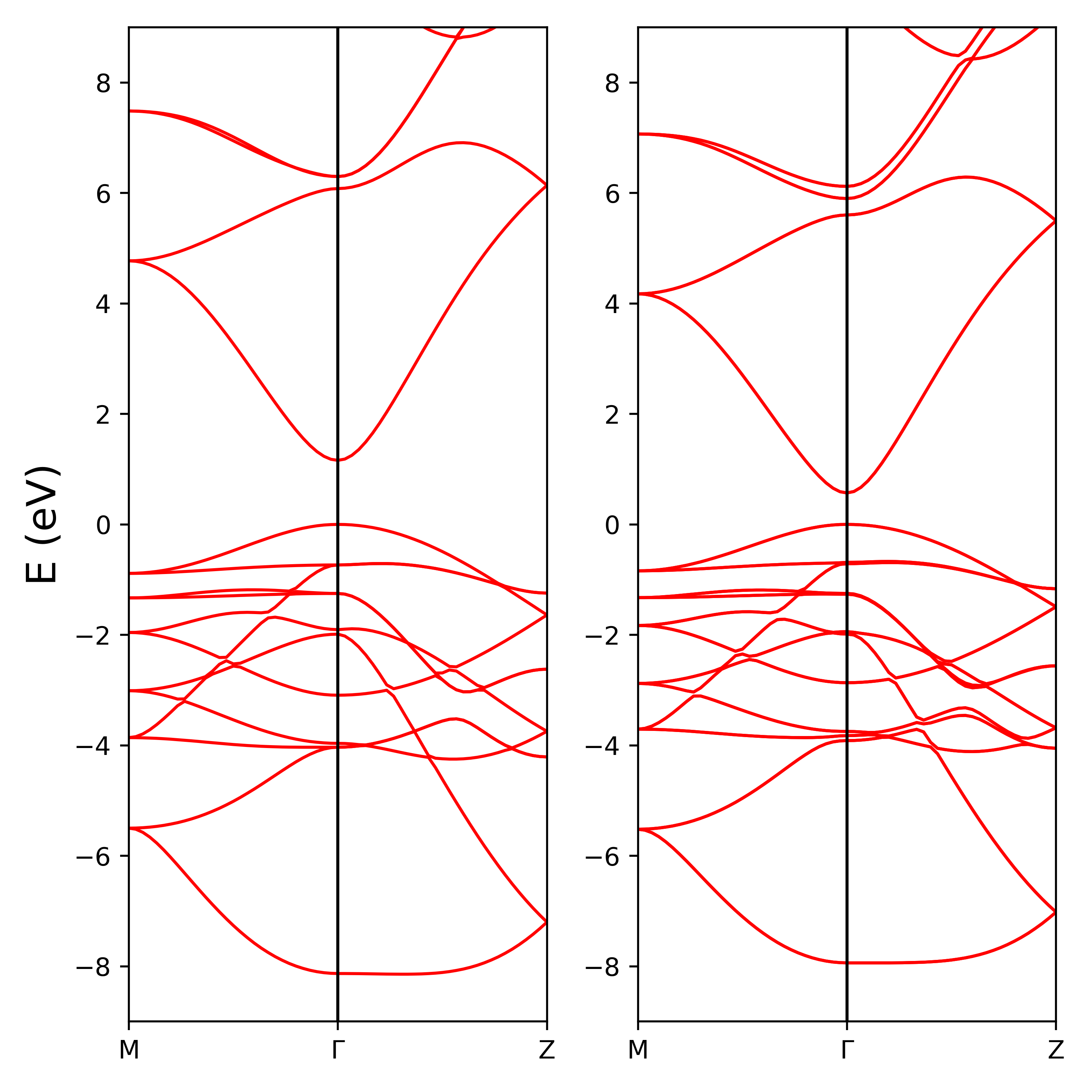
在加入SOC之后,计算绝缘体价带数等于赝势中价电子个数,否则是价电子个数的一半,例如$SnO_{2}$,计算中的价带个数,Sn考虑了s、p、d价电子14个,O考虑了6个价电子,赝势中价电子数是UPF文件中的z_valence值。原胞中2个Sn原子、4个O原子,一共14*2+6*4=52个价电子,在SOC计算中价带顶是第52个带,而在非SOC计算中价带顶是第26个带。
不支持relax计算(QE未实现)。
Spin-orbit只对较重元素具有明显的效应。参考标准从第5周期元素开始加入自旋轨道耦合。
以$SnO_{2}$为例,用不加SOC做vc-relax得到晶格常数结果(可以换算得到celldm(1)=9.145886),进行SOC计算。
&CONTROL
calculation='scf',
disk_io='low',
prefix='pwscf',
pseudo_dir='./',
outdir='./tmp',
verbosity='high'
tprnfor=.true.
tstress=.true.
forc_conv_thr=1.0d-5
/
&SYSTEM
ibrav= 6,
celldm(1)=9.145886,
celldm(3)=0.6713850,
nat= 6,
ntyp= 2,
occupations = 'smearing',
smearing = 'gauss',
degauss = 1.0d-9
ecutwfc = 50
ecutrho = 500,
noncolin=.true., lspinorb=.true.,
/
&ELECTRONS
electron_maxstep = 500
conv_thr = 1.0d-10
mixing_beta = 0.7d0
diagonalization = 'david'
/
&IONS
ion_dynamics='bfgs'
/
&CELL
/
ATOMIC_SPECIES
Sn 118.710 Sn.rel-pbe-dn-rrkjus_psl.1.0.0.UPF
O 15.999 O.rel-pbe-n-rrkjus_psl.1.0.0.UPF
K_POINTS {automatic}
6 6 9 0 0 0
ATOMIC_POSITIONS (crystal)
Sn 0.000000000 0.000000000 -0.000000000
Sn 0.500000000 0.500000000 0.500000000
O 0.306642111 0.306642111 -0.000000000
O -0.306642111 -0.306642111 -0.000000000
O 0.193357889 0.806642111 0.500000000
O 0.806642111 0.193357889 0.500000000
做能带计算输入如下:
&CONTROL
calculation='bands',
disk_io='low',
prefix='pwscf',
pseudo_dir='./',
outdir='./tmp',
verbosity='high'
tprnfor=.true.
tstress=.true.
forc_conv_thr=1.0d-5
/
&SYSTEM
ibrav= 6,
celldm(1)=9.145886,
celldm(3)=0.6713850,
nat= 6,
ntyp= 2,
occupations = 'smearing',
smearing = 'gauss',
degauss = 1.0d-9
ecutwfc = 50
ecutrho = 500,
noncolin=.true., lspinorb=.true.,
/
&ELECTRONS
electron_maxstep = 500
conv_thr = 1.0d-10
mixing_beta = 0.7d0
diagonalization = 'david'
/
&IONS
ion_dynamics='bfgs'
/
&CELL
/
ATOMIC_SPECIES
Sn 118.710 Sn.rel-pbe-dn-rrkjus_psl.1.0.0.UPF
O 15.999 O.rel-pbe-n-rrkjus_psl.1.0.0.UPF
ATOMIC_POSITIONS (crystal)
Sn 0.000000000 0.000000000 -0.000000000
Sn 0.500000000 0.500000000 0.500000000
O 0.306642111 0.306642111 -0.000000000
O -0.306642111 -0.306642111 -0.000000000
O 0.193357889 0.806642111 0.500000000
O 0.806642111 0.193357889 0.500000000
K_POINTS {crystal_b}
3
M 30
gG 30
Z 0
结果如图所示,左侧是未加入SOC的能带,右侧是加了SOC的能带。大家一定可以找到有些自旋轨道耦合造成分裂的带。关于自旋轨道耦合完整的理论分析,参见文献[14]。
7.2 自旋极化
基于局域自旋密度近似(LSDA)。
输入中加入nspin = 2。设置至少一种元素的starting_magnetization(i), i=1…ntyp值非零,scf计算中磁矩从此值逐渐减小直到收敛(只减不增,通常设为1)。
自旋极化不必要full relativistic赝势(见7.3节),支持relax计算。
7.2.1 同种元素不同磁矩的设置
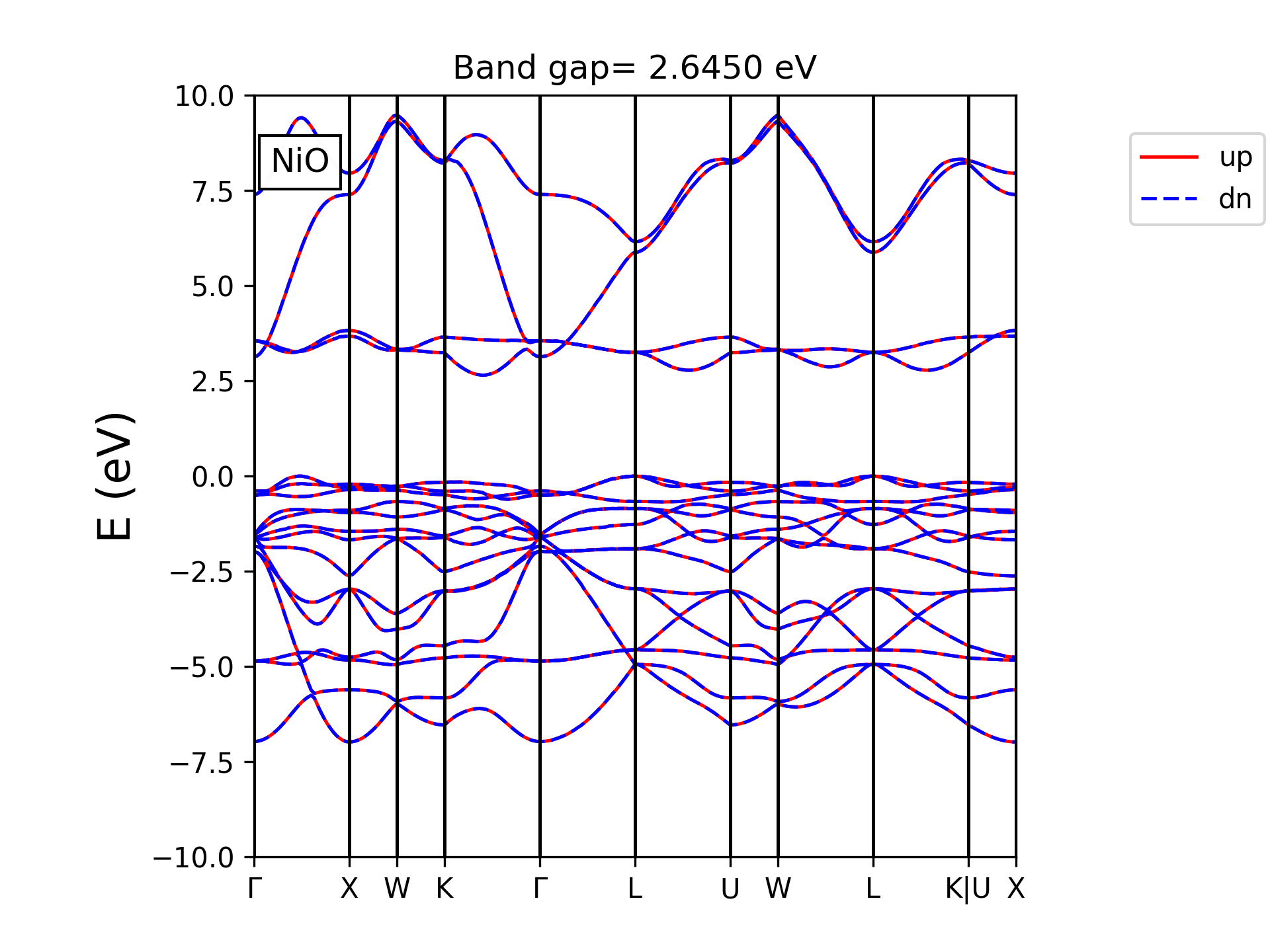
例:反铁磁NiO。
&control
calculation='scf', disk_io='low', prefix='pwscf',
pseudo_dir='./', outdir='./tmp', verbosity='high'
/
&system
ibrav= 0, celldm(1)=7.93, nat= 4, ntyp= 3,
ecutwfc = 50.0, ecutrho = 500.0,
occupations='smearing', smearing='mv', degauss=0.02,
nspin=2,
starting_magnetization(2)= 0.5,
starting_magnetization(3)=-0.5,
/
&electrons
conv_thr = 1.0d-9
mixing_beta = 0.8d0
diagonalization = 'david'
/
CELL_PARAMETERS (alat)
0.50 0.50 1.00
0.50 1.00 0.50
1.00 0.50 0.50
ATOMIC_SPECIES
O 16.00 O.UPF
Ni1 58.69 Ni.UPF
Ni2 58.69 Ni.UPF
ATOMIC_POSITIONS (crystal)
O 0.25 0.25 0.25
O 0.75 0.75 0.75
Ni1 0.00 0.00 0.00
Ni2 0.50 0.50 0.50
K_POINTS (automatic)
6 6 6 0 0 0
计算输出的结尾可以找到:
total magnetization = 0.00 Bohr mag/cell
absolute magnetization = 2.87 Bohr mag/cell
总磁矩是0.00,与反铁磁符合(此材料需要LDA+U或Hybrid functional以得到带隙)。
7.3 非共线磁性
同时加入了自旋极化和非共线自旋。
在输入中加入noncolin=.true., lspinorb=.true.,(这时不要设置nspin)。设置至少一种元素的starting_magnetization(i)值非零, i=1…ntyp。设置angle1(i),angle2(i)。使用至少一种full relativistic赝势。
8. Meta-GGA计算
QE中对于meta-gga的计算通常需要安装libxc,但也有例外,如TPSS,M06-L不需要libxc。
8.1 用Meta-GGA计算过渡金属氧化物
8.1.1 编译qe的libxc版本
下载并解压libxc-4.3.4.tar.gz。在解压目录运行:
./configure --prefix=/home/username/local
make
make install
其中/home/username/local换成实际目录,make install后会生成libxc相关的库文件及头文件在这个目录。
下载并解压QE6.5,在解压目录运行:
./configure --with-libxc --with-libxc-prefix=/home/username/local --with-libxc-include=/home/username/local/include
修改make.inc,加入-lxcf03 ,注意顺序
LIBXC_LIBS = -L/home/username/local/lib -lxcf90 -lxcf03 -lxc
编译QE
make pw pp
8.1.2 用SCAN计算氧化物的能带
首先进行scf计算,目前meta-GGA计算仅支持模守恒赝势。
&control
calculation='scf', disk_io='low', prefix='pwscf',
pseudo_dir='./', outdir='./tmp', verbosity='high'
/
&system
ibrav= 0, celldm(1)=7.93, nat= 4, ntyp= 3,
ecutwfc = 50.0, ecutrho = 500.0,
occupations='smearing', smearing='mv', degauss=1.0d-2,
nspin=2,
starting_magnetization(2)= 0.5,
starting_magnetization(3)=-0.5,
input_dft='scan'
/
&electrons
conv_thr = 1.0d-9
mixing_beta = 0.8d0
diagonalization = 'david'
/
CELL_PARAMETERS (alat)
0.50 0.50 1.00
0.50 1.00 0.50
1.00 0.50 0.50
ATOMIC_SPECIES
O 16.00 O.UPF
Ni1 58.69 Ni.UPF
Ni2 58.69 Ni.UPF
ATOMIC_POSITIONS (crystal)
O 0.25 0.25 0.25
O 0.75 0.75 0.75
Ni1 0.00 0.00 0.00
Ni2 0.50 0.50 0.50
K_POINTS (automatic)
6 6 6 0 0 0
接着做nscf的能带计算。设置calculation='bands',将$\mathbf{k}$点设置如下:
K_POINTS (crystal_b)
12
0.0 0.0 0.0 30 !G
0.5 0.0 0.5 15 !X
0.5 0.25 0.75 15 !W
0.375 0.375 0.75 30 !K
0.0 0.0 0.0 30 !G
0.5 0.5 0.5 30 !L
0.625 0.25 0.625 15 !U
0.5 0.25 0.75 30 !W
0.5 0.5 0.5 30 !L
0.375 0.375 0.75 0 !K
0.625 0.25 0.625 15 !U
0.5 0.0 0.5 1 !X
接着做bands.x计算,因为有两个自旋分量,需要做两次,输入分别为:
&bands
prefix='pwscf',
outdir='tmp'
filband='bd1.dat'
spin_component=1
lp=.true.
/
&bands
prefix='pwscf',
outdir='tmp'
filband='bd2.dat'
spin_component=2
lp=.true.
/
bands.x计算在目前版本(6.5)还有一个小问题,即出现如下报错:
Reading xml data from directory:
tmp/pwscf.save/
Message from routine volume:
axis vectors are left-handed
%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%
Error in routine set_dft_from_name (1):
XC-000-000-000-000: unrecognized dft
%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%
解决办法是在tmp/pwscf.xml和tmp/pwscf.save/data-file-schema.xml中找到:
<functional>XC-000-000-000-000</functional>
改成:
<functional>scan</functional>
或使用命令:
sed -i 's/XC-000-000-000-000/scan/g' tmp/pwscf.xml
sed -i 's/XC-000-000-000-000/scan/g' tmp/pwscf.save/data-file-schema.xml
计算结果画图如下,与7.2.1节PBE的计算结果没有带隙相比,描述反铁磁过渡金属氧化物的能带有所提升,这里的SCAN计算没有引入经验参数[18],从计算结果上看,SCAN这种交换关联泛函对不同材料体系的描述比PBE更具有普遍性,同时SCAN的计算也较TPSS更容易收敛,有望成为新的基准交换关联泛函。
本节计算的输入、输出文件见这里。
8.2 用TB-mBJ修正带隙
查看此内容(收费99元),欢迎发邮件至yyyu200@163.com。
9. DFT+U计算
9.1 DFT+U计算
9.2 使用HP模块的DFT+U计算
10. Raman及红外光谱计算
步骤同4.1节单个q点的ph.x计算。参考安装包例子q-e-qe-7.0/PHonon/examples/example05和example15。
11. EPR和NMR化学位移计算
References
-
https://avogadro.cc/docs/building-materials/reducing-crystals-to-primitive-cells/
-
https://www.researchgate.net/post/How_to_convert_a_conventional_cell_to_a_primitive_cell
-
Bilbao, http://www.cryst.ehu.es/cryst/celltran.html
-
Wahyu Setyawan, Stefano Curtarolo, High-throughput electronic band structure calculations: Challenges and tools. Computational Materials Science 49 (2010) 299–312.(doi:10.1016/j.commatsci.2010.05.010).
-
http://www.ioffe.ru/SVA/NSM/Semicond/SiC/bandstr.html
-
Sgiarovello, C., Binggeli, N., Baldereschi, A. (2004). Surface morphology and ionization potentials of polar semiconductors: The case of GaAs. Physical Review B, 69(3). doi:10.1103/PhysRevB.69.035320.
-
F. Bassani, G. Parravicini, Electronic states and optical transitions in solids. Pergamon Press (1975).
-
Anna Tararan et al. Optical gap and optically active intragap defects in cubic BN. Phys. Rev. B 98, 094106 (2018).
-
Martin Dressel, George Gruner, Electrodynamics of Solids-Optical Properties of Electrons in Matter. Cambridge University Press (2002).
-
P. Giannozzi et al, Ab initio calculation of phonon dispersions in semiconductors. Phys. Rev. B 43, 7231 (1991).. L. Lindsay et al, Ab initio thermal transport in compound semiconductors. Physical Review B 87,165201 (2013).
-
P.Y. Yu, M. Cardona, Fundamentals of Semiconductors: Physics and Materials Properties, Springer, 2010.
-
J. Paier et al, Screened hybrid density functionals applied to solids, The Journal of Chemical Physics 124, 154709 (2006); doi: 10.1063/1.2187006.
-
Marzari et al, Maximally localized Wannier functions: Theory and applications, Rev. Mod. Phys. 84, 1419 (2012).
-
X Zhang, Q Liu, JW Luo, AJ Freeman, A Zunger. Hidden spin polarization in inversion-symmetric bulk crystals. Nature Physics 10 (5), 387-393(2014).
-
I. Timrov, N. Vast, R. Gebauer, S. Baroni, turboEELS — A code for the simulation of the electron energy loss and inelastic X-ray scattering spectra using the Liouville–Lanczos approach to time-dependent density-functional perturbation theory, Computer Physics Communications (2015).
-
Iurii Timrov, Maxime Markov, Tommaso Gorni, Michèle Raynaud, Oleksandr Motornyi, Ralph Gebauer, Stefano Baroni, and Nathalie Vast. Ab initio study of electron energy loss spectra of bulk bismuth up to 100 eV. Phys. Rev. B 95, 094301 (2017).
-
Onida, Giovanni and Reining, Lucia and Rubio, Angel. Electronic excitations: density-functional versus many-body Green’s-function approaches. Rev. Mod. Phys., 74, 601(2002).
-
Christopher Lane, James W. Furness, Ioana Gianina Buda, Yubo Zhang, Robert S. Markiewicz, Bernardo Barbiellini, Jianwei Sun, and Arun Bansil, Antiferromagnetic ground state of La2CuO4: A parameter-free ab initio description. Phys. Rev. B 98, 125140(2018).
-
Ivan Carnimeo, Stefano Baroni and Paolo Giannozzi, Fast hybrid density-functional computations using plane-wave basis sets. Electron. Struct. 1,015009(2019).
Update On 2019/09/05
Update On 2020/01/22
Update On 2020/05/03
Update On 2020/07/01
本文总阅读量次